

A Single CRISPR-Cas9 Deletion Strategy that Targets the Majority of DMD Patients Restores Dystrophin Function in hiPSC-Derived Muscle Cells
- PMID: 26877224
- PMCID: PMC4826286
- DOI: 10.1016/j.stem.2016.01.021
A Single CRISPR-Cas9 Deletion Strategy that Targets the Majority of DMD Patients Restores Dystrophin Function in hiPSC-Derived Muscle Cells
Abstract
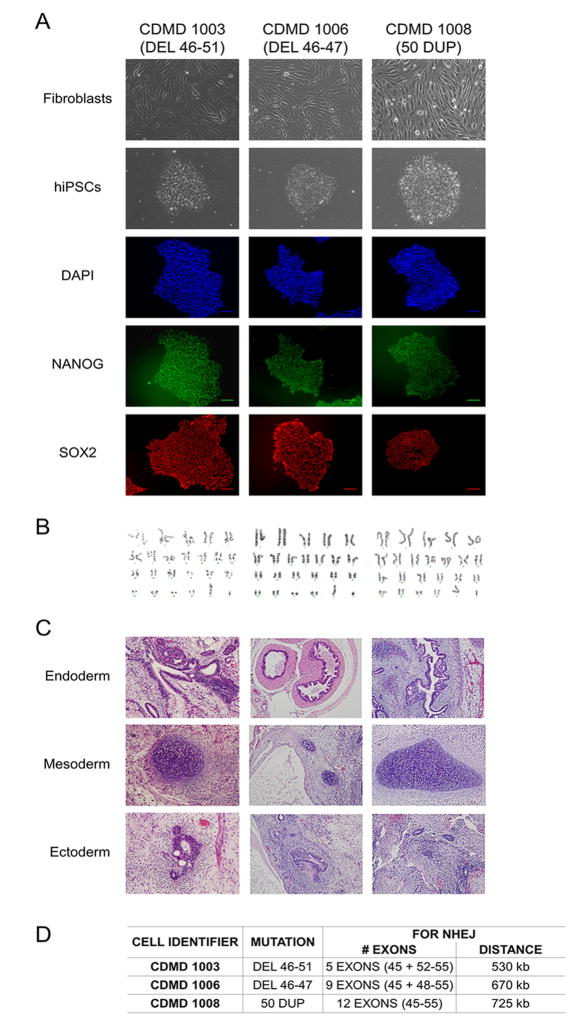
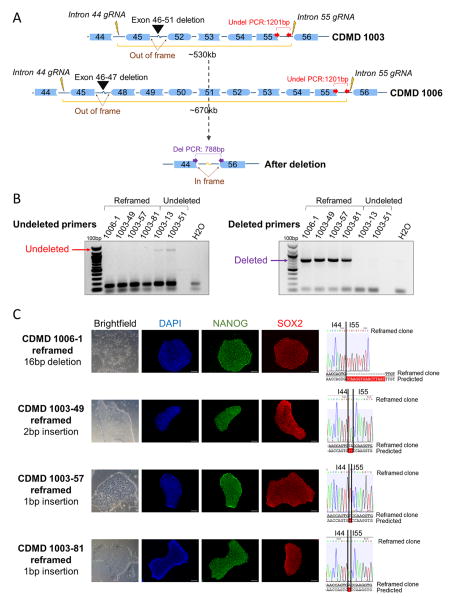
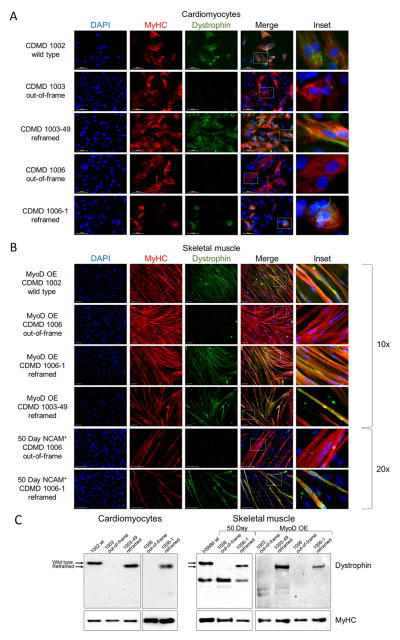
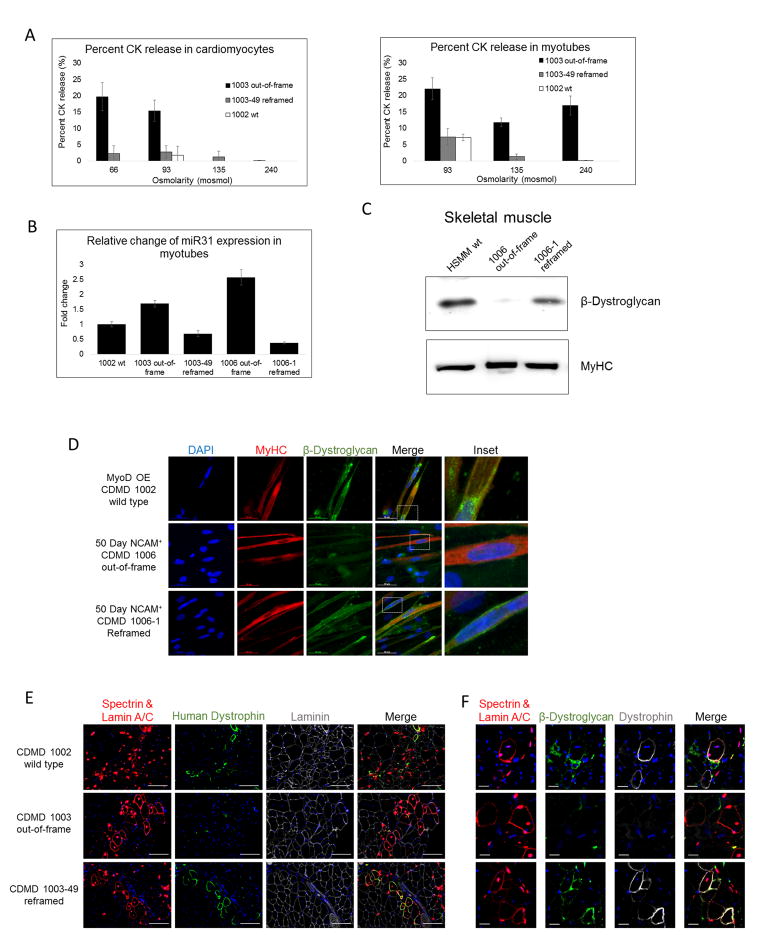
Mutations in DMD disrupt the reading frame, prevent dystrophin translation, and cause Duchenne muscular dystrophy (DMD). Here we describe a CRISPR/Cas9 platform applicable to 60% of DMD patient mutations. We applied the platform to DMD-derived hiPSCs where successful deletion and non-homologous end joining of up to 725 kb reframed the DMD gene. This is the largest CRISPR/Cas9-mediated deletion shown to date in DMD. Use of hiPSCs allowed evaluation of dystrophin in disease-relevant cell types. Cardiomyocytes and skeletal muscle myotubes derived from reframed hiPSC clonal lines had restored dystrophin protein. The internally deleted dystrophin was functional as demonstrated by improved membrane integrity and restoration of the dystrophin glycoprotein complex in vitro and in vivo. Furthermore, miR31 was reduced upon reframing, similar to observations in Becker muscular dystrophy. This work demonstrates the feasibility of using a single CRISPR pair to correct the reading frame for the majority of DMD patients.
Copyright © 2016 Elsevier Inc. All rights reserved.
Figures
Comment in
-
Imprecision Medicine: A One-Size-Fits-Many Approach for Muscle Dystrophy.Cell Stem Cell. 2016 Apr 7;18(4):423-4. doi: 10.1016/j.stem.2016.03.004. Cell Stem Cell. 2016. PMID: 27058929
Similar articles
-
Creation of a Novel Humanized Dystrophic Mouse Model of Duchenne Muscular Dystrophy and Application of a CRISPR/Cas9 Gene Editing Therapy.J Neuromuscul Dis. 2017;4(2):139-145. doi: 10.3233/JND-170218. J Neuromuscul Dis. 2017. PMID: 28505980 Free PMC article.
-
Correction of Three Prominent Mutations in Mouse and Human Models of Duchenne Muscular Dystrophy by Single-Cut Genome Editing.Mol Ther. 2020 Sep 2;28(9):2044-2055. doi: 10.1016/j.ymthe.2020.05.024. Epub 2020 May 30. Mol Ther. 2020. PMID: 32892813 Free PMC article.
-
Molecular correction of Duchenne muscular dystrophy by splice modulation and gene editing.RNA Biol. 2021 Jul;18(7):1048-1062. doi: 10.1080/15476286.2021.1874161. Epub 2021 Jan 20. RNA Biol. 2021. PMID: 33472516 Free PMC article. Review.
-
In vivo genome editing in mouse restores dystrophin expression in Duchenne muscular dystrophy patient muscle fibers.Genome Med. 2021 Apr 12;13(1):57. doi: 10.1186/s13073-021-00876-0. Genome Med. 2021. PMID: 33845891 Free PMC article.
-
Therapeutic Applications of CRISPR/Cas for Duchenne Muscular Dystrophy.Curr Gene Ther. 2017;17(4):301-308. doi: 10.2174/1566523217666171121165046. Curr Gene Ther. 2017. PMID: 29173172 Review.
Cited by
-
Multiple Exon Skipping in the Duchenne Muscular Dystrophy Hot Spots: Prospects and Challenges.J Pers Med. 2018 Dec 7;8(4):41. doi: 10.3390/jpm8040041. J Pers Med. 2018. PMID: 30544634 Free PMC article. Review.
-
CRISPR-Cas9-Mediated Gene Therapy in Neurological Disorders.Mol Neurobiol. 2022 Feb;59(2):968-982. doi: 10.1007/s12035-021-02638-w. Epub 2021 Nov 23. Mol Neurobiol. 2022. PMID: 34813019 Review.
-
Biomaterials-mediated CRISPR/Cas9 delivery: recent challenges and opportunities in gene therapy.Front Chem. 2023 Sep 28;11:1259435. doi: 10.3389/fchem.2023.1259435. eCollection 2023. Front Chem. 2023. PMID: 37841202 Free PMC article. Review.
-
Recapitulating human myogenesis ex vivo using human pluripotent stem cells.Exp Cell Res. 2022 Feb 15;411(2):112990. doi: 10.1016/j.yexcr.2021.112990. Epub 2021 Dec 30. Exp Cell Res. 2022. PMID: 34973262 Free PMC article. Review.
-
Repairing folding-defective α-sarcoglycan mutants by CFTR correctors, a potential therapy for limb-girdle muscular dystrophy 2D.Hum Mol Genet. 2018 Mar 15;27(6):969-984. doi: 10.1093/hmg/ddy013. Hum Mol Genet. 2018. PMID: 29351619 Free PMC article.
References
-
- Arechavala-Gomeza V, Anthony K, Morgan J, Muntoni F. Antisense Oligonucleotide-Mediated Exon Skipping for Duchenne Muscular Dystrophy: Progress and Challenges. Curr Gene Ther. 2012;12:152–160. - PubMed
-
- Beroud C, Tuffery-Giraud S, Matsuo M, Hamroun D, Calemard LM, Boisseau P, Blayau M, Philippe C, Cosse M, Page M, et al. Multiexon skipping leading to an artificial DMD protein lacking amino acids from exons 45 through 55 could rescue up to 63% of patients with Duchenne muscular dystrophy. Hum Mutat. 2007;28:196–202. - PubMed
-
- Carsana A, Frisso G, Tremolaterra M, Lanzillo R, Vitale D, Santoro L, Salvatore F. Analysis of dystrophin gene deletions indicates that the hinge III region of the protein correlates with disease severity. Ann Hum Genet. 2005;69:253–9. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
