

Blocking mitochondrial calcium release in Schwann cells prevents demyelinating neuropathies
- PMID: 26878172
- PMCID: PMC4767343
- DOI: 10.1172/JCI84505
Blocking mitochondrial calcium release in Schwann cells prevents demyelinating neuropathies
Erratum in
-
Blocking mitochondrial calcium release in Schwann cells prevents demyelinating neuropathies.J Clin Invest. 2016 Apr 1;126(4):1605. doi: 10.1172/JCI87203. Epub 2016 Mar 7. J Clin Invest. 2016. PMID: 26950425 Free PMC article. No abstract available.
Retraction in
-
Blocking mitochondrial calcium release in Schwann cells prevents demyelinating neuropathies.J Clin Invest. 2017 Mar 1;127(3):1115. doi: 10.1172/JCI92100. Epub 2017 Mar 1. J Clin Invest. 2017. PMID: 28248206 Free PMC article. No abstract available.
Expression of concern in
-
Blocking mitochondrial calcium release in Schwann cells prevents demyelinating neuropathies.J Clin Invest. 2016 Jul 1;126(7):2773. doi: 10.1172/JCI88179. Epub 2016 Jul 1. J Clin Invest. 2016. PMID: 27111236 Free PMC article. No abstract available.
Abstract
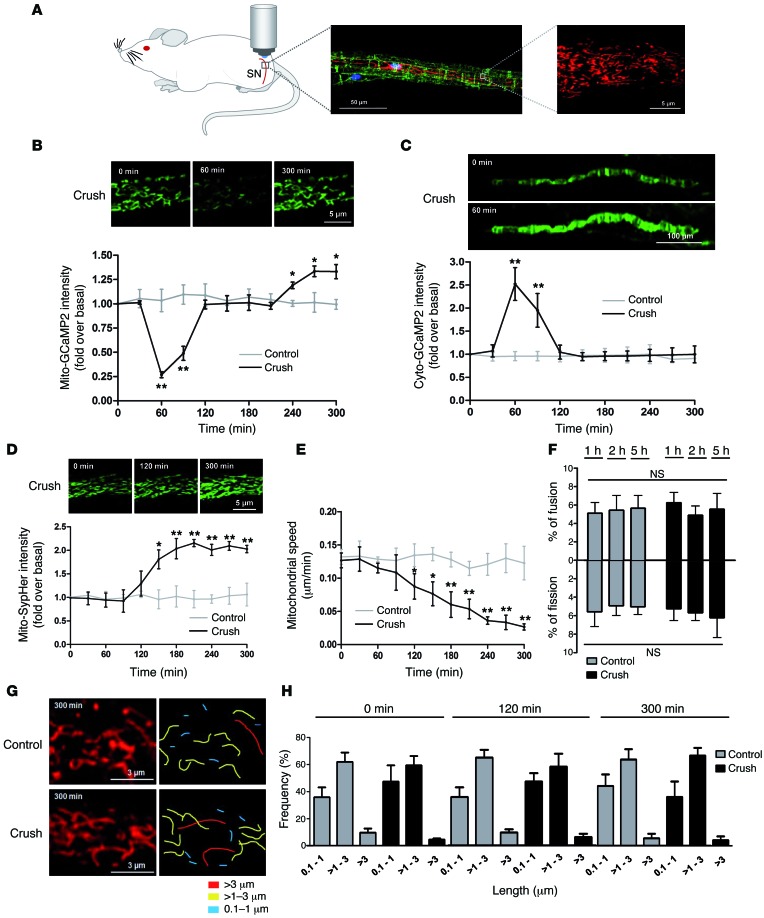
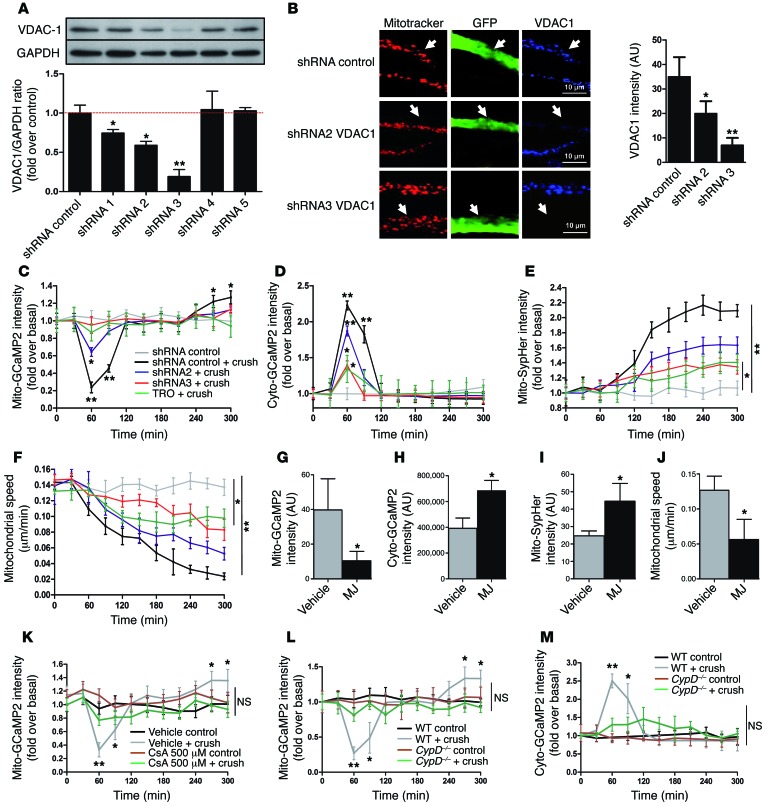
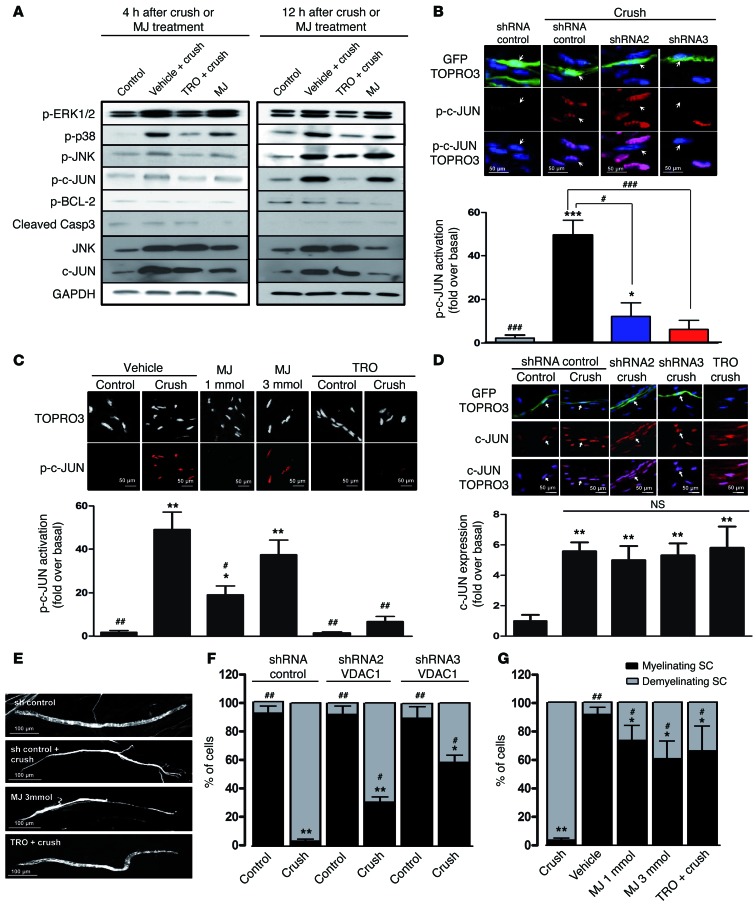
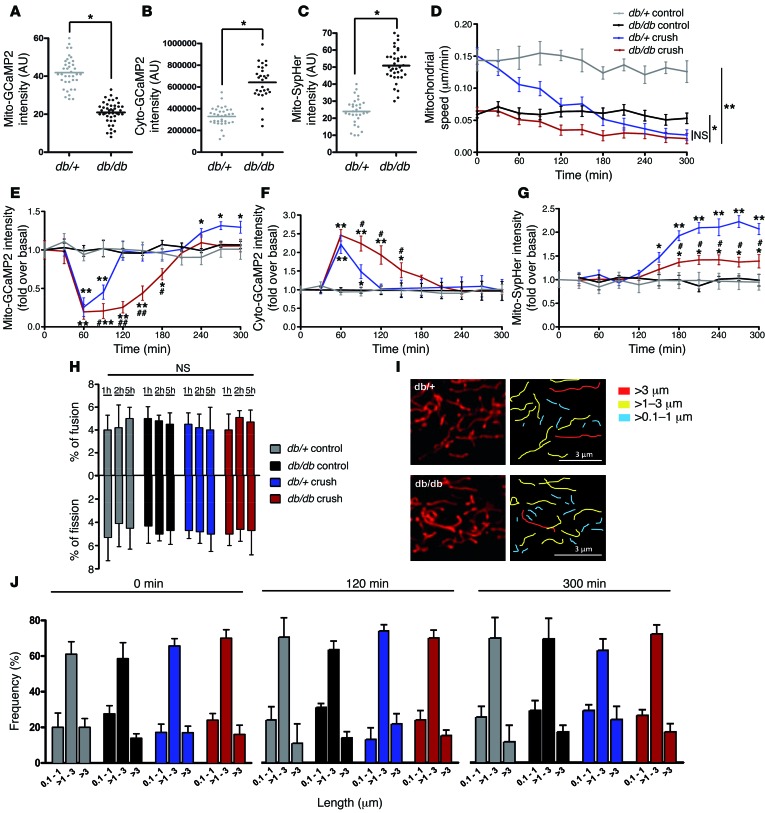
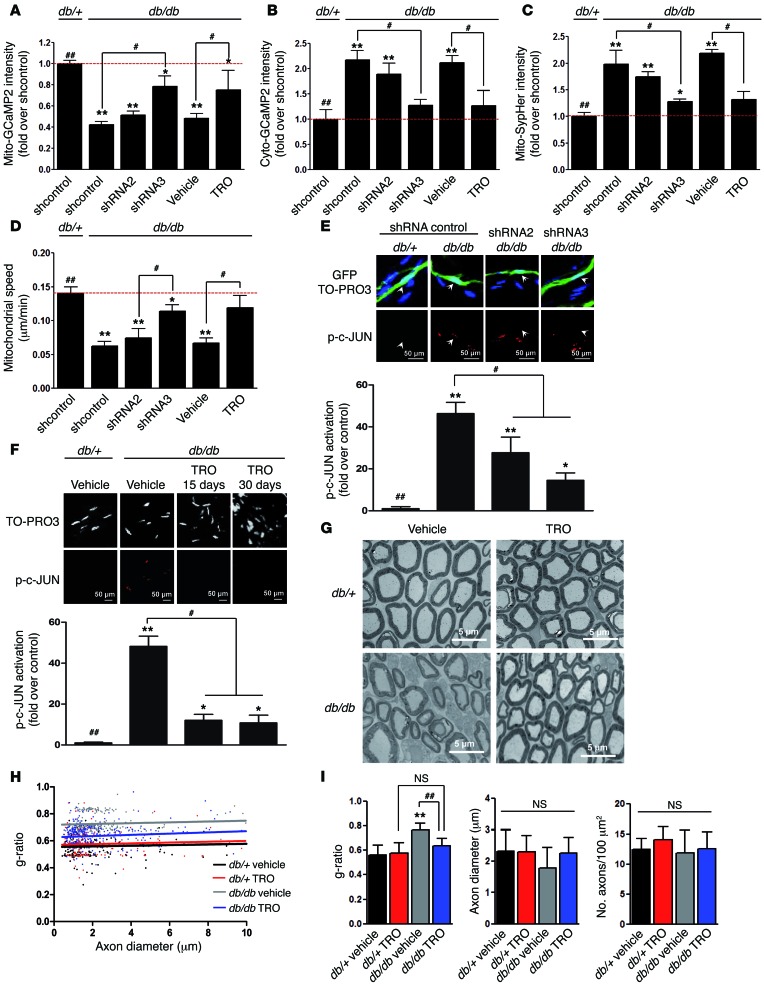
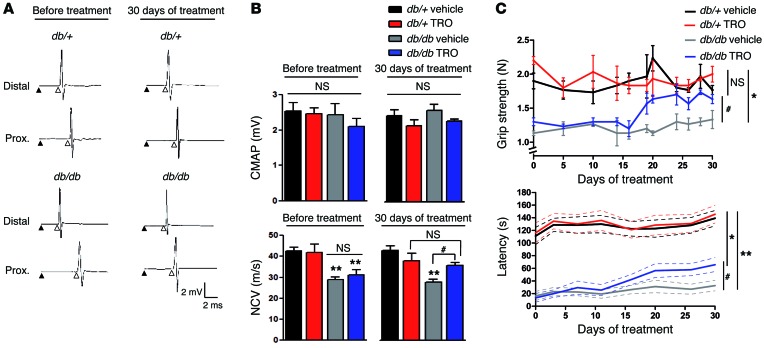
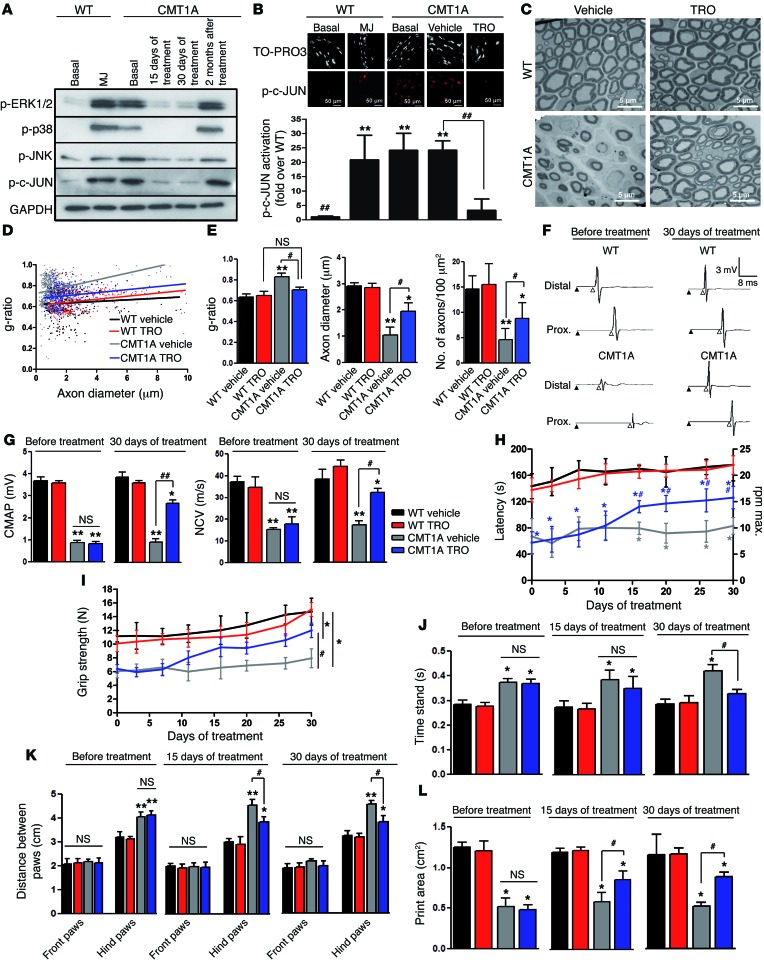
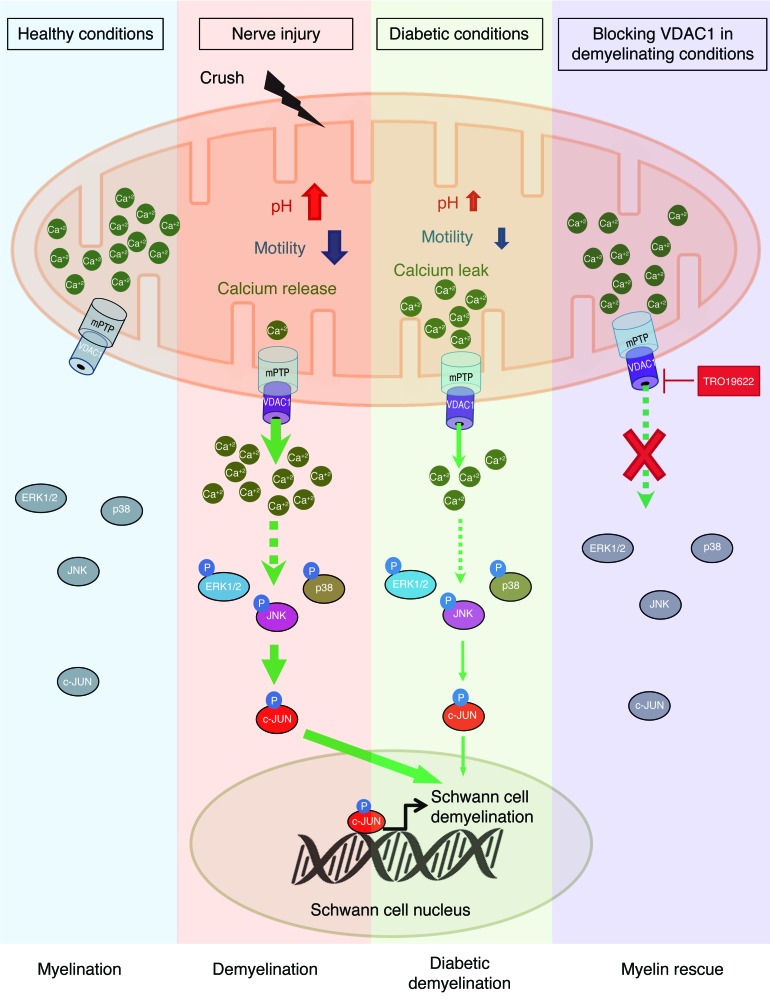
Schwann cells produce myelin sheath around peripheral nerve axons. Myelination is critical for rapid propagation of action potentials, as illustrated by the large number of acquired and hereditary peripheral neuropathies, such as diabetic neuropathy or Charcot-Marie-Tooth diseases, that are commonly associated with a process of demyelination. However, the early molecular events that trigger the demyelination program in these diseases remain unknown. Here, we used virally delivered fluorescent probes and in vivo time-lapse imaging in a mouse model of demyelination to investigate the underlying mechanisms of the demyelination process. We demonstrated that mitochondrial calcium released by voltage-dependent anion channel 1 (VDAC1) after sciatic nerve injury triggers Schwann cell demyelination via ERK1/2, p38, JNK, and c-JUN activation. In diabetic mice, VDAC1 activity was altered, resulting in a mitochondrial calcium leak in Schwann cell cytoplasm, thereby priming the cell for demyelination. Moreover, reduction of mitochondrial calcium release, either by shRNA-mediated VDAC1 silencing or pharmacological inhibition, prevented demyelination, leading to nerve conduction and neuromuscular performance recovery in rodent models of diabetic neuropathy and Charcot-Marie-Tooth diseases. Therefore, this study identifies mitochondria as the early key factor in the molecular mechanism of peripheral demyelination and opens a potential opportunity for the treatment of demyelinating peripheral neuropathies.
Figures
Similar articles
-
Desert hedgehog is a mediator of demyelination in compression neuropathies.Exp Neurol. 2015 Sep;271:84-94. doi: 10.1016/j.expneurol.2015.04.014. Epub 2015 May 1. Exp Neurol. 2015. PMID: 25936873
-
Myelin abnormality in Charcot-Marie-Tooth type 4J recapitulates features of acquired demyelination.Ann Neurol. 2018 Apr;83(4):756-770. doi: 10.1002/ana.25198. Epub 2018 Mar 30. Ann Neurol. 2018. PMID: 29518270 Free PMC article.
-
Targeting the colony stimulating factor 1 receptor alleviates two forms of Charcot-Marie-Tooth disease in mice.Brain. 2015 Nov;138(Pt 11):3193-205. doi: 10.1093/brain/awv240. Epub 2015 Aug 21. Brain. 2015. PMID: 26297559
-
Schwann cells and the pathogenesis of inherited motor and sensory neuropathies (Charcot-Marie-Tooth disease).Glia. 2006 Sep;54(4):243-57. doi: 10.1002/glia.20386. Glia. 2006. PMID: 16856148 Review.
-
Genetic mechanisms of peripheral nerve disease.Neurosci Lett. 2021 Jan 18;742:135357. doi: 10.1016/j.neulet.2020.135357. Epub 2020 Nov 26. Neurosci Lett. 2021. PMID: 33249104 Review.
Cited by
-
Peripheral Glial Cells in the Development of Diabetic Neuropathy.Front Neurol. 2018 May 2;9:268. doi: 10.3389/fneur.2018.00268. eCollection 2018. Front Neurol. 2018. PMID: 29770116 Free PMC article. Review.
-
Axon degeneration: make the Schwann cell great again.Neural Regen Res. 2017 Apr;12(4):518-524. doi: 10.4103/1673-5374.205000. Neural Regen Res. 2017. PMID: 28553320 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
