

Initial Suppression of Transforming Growth Factor-β Signaling and Loss of TGFBI Causes Early Alveolar Structural Defects Resulting in Bronchopulmonary Dysplasia
- PMID: 26878215
- PMCID: PMC5808152
- DOI: 10.1016/j.ajpath.2015.11.024
Initial Suppression of Transforming Growth Factor-β Signaling and Loss of TGFBI Causes Early Alveolar Structural Defects Resulting in Bronchopulmonary Dysplasia
Abstract
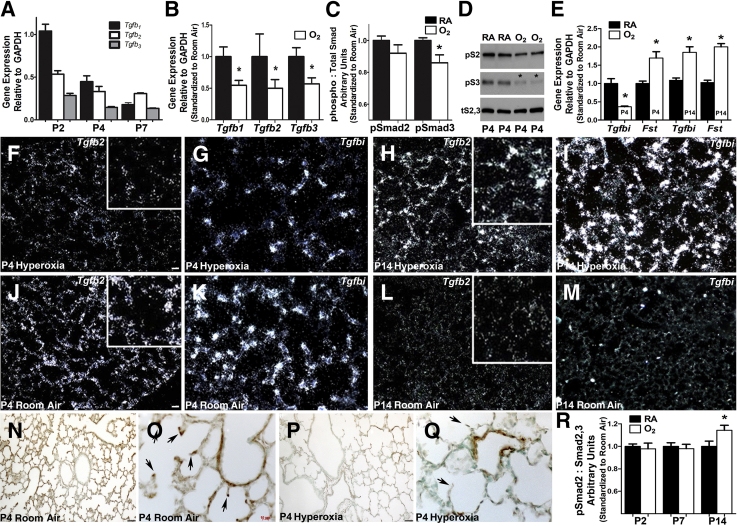
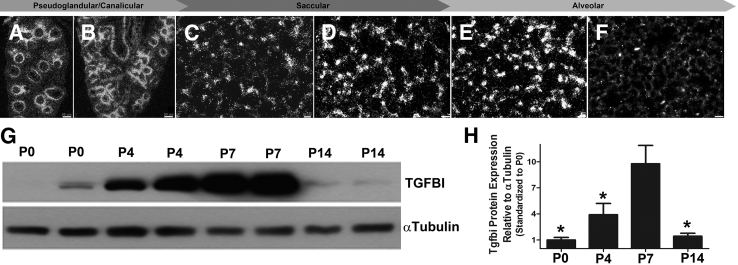
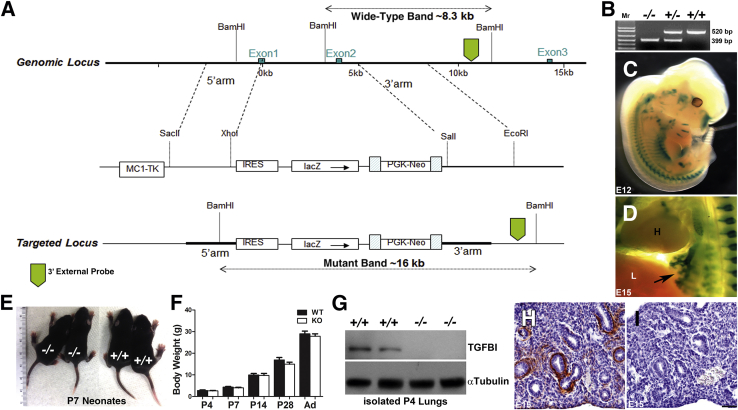
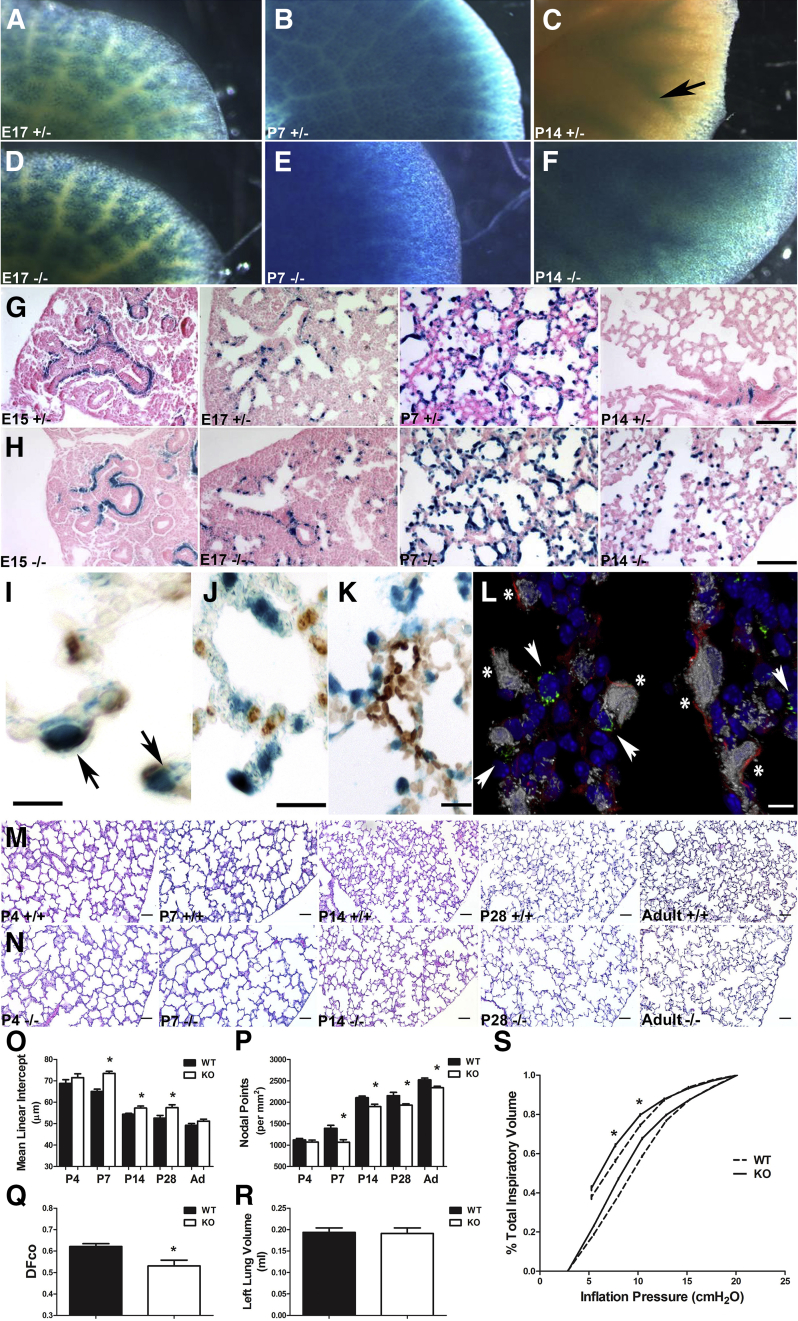
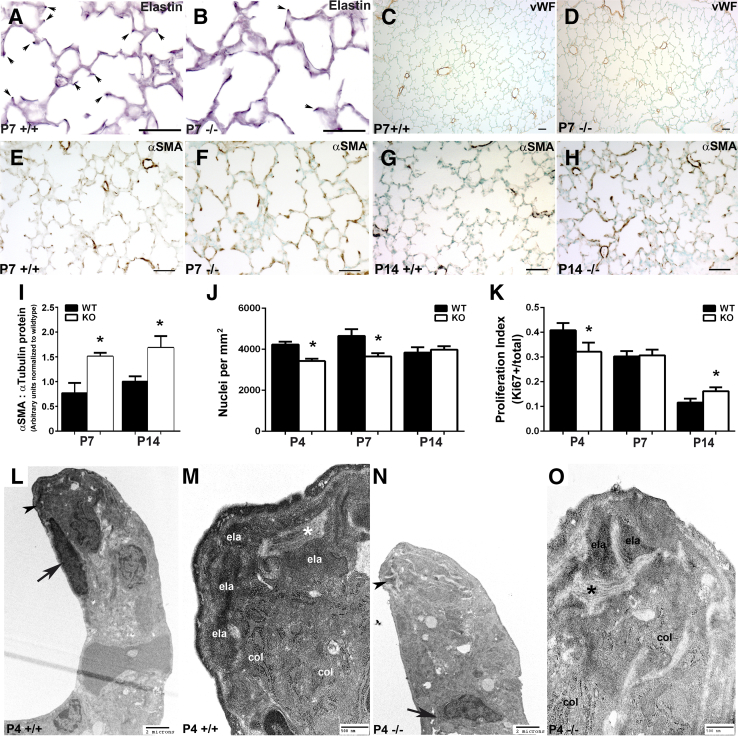
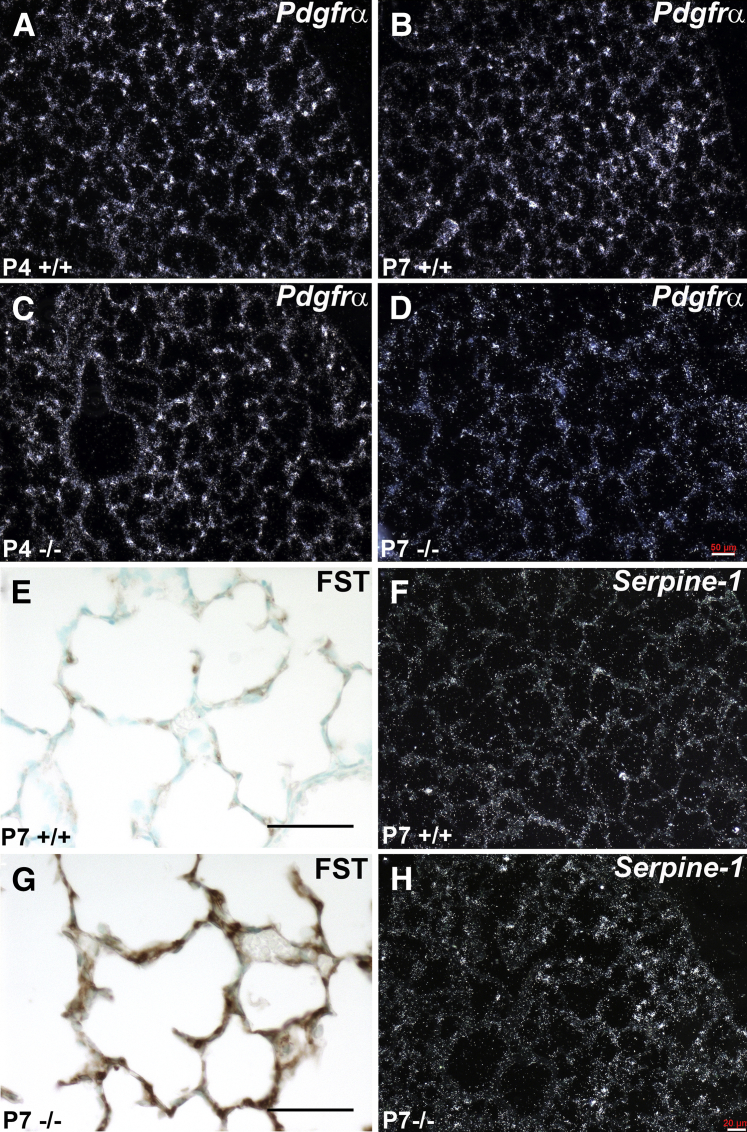
Septation of the gas-exchange saccules of the morphologically immature mouse lung requires regulated timing, spatial direction, and dosage of transforming growth factor (TGF)-β signaling. We found that neonatal hyperoxia acutely initially diminished saccular TGF-β signaling coincident with alveolar simplification. However, sustained hyperoxia resulted in a biphasic response and subsequent up-regulation of TGF-β signaling, ultimately resulting in bronchopulmonary dysplasia. Significantly, we found that the TGF-β-induced matricellular protein (TGFBI) was similarly biphasically altered in response to hyperoxia. Moreover, genetic ablation revealed that TGFBI was required for normal alveolar structure and function. Although the phenotype was not neonatal lethal, Tgfbi-deficient lungs were morphologically abnormal. Mutant septal tips were stunted, lacked elastin-positive tips, exhibited reduced proliferation, and contained abnormally persistent alveolar α-smooth muscle actin myofibroblasts. In addition, Tgfbi-deficient lungs misexpressed TGF-β-responsive follistatin and serpine 1, and transiently suppressed myofibroblast platelet-derived growth factor α differentiation marker. Finally, despite normal lung volume, Tgfbi-null lungs displayed diminished elastic recoil and gas exchange efficiency. Combined, these data demonstrate that initial suppression of the TGF-β signaling apparatus, as well as loss of key TGF-β effectors (like TGFBI), underlies early alveolar structural defects, as well as long-lasting functional deficits routinely observed in chronic lung disease of infancy patients. These studies underline the complex (and often contradictory) role of TGF-β and indicate a need to design studies to associate alterations with initial appearance of phenotypical changes suggestive of bronchopulmonary dysplasia.
Copyright © 2016 American Society for Investigative Pathology. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Kimura J., Deutsch G.H. Key mechanisms of early lung development. Pediatr Dev Pathol. 2007;10:335–347. - PubMed
-
- Burri P.H. Fetal and postnatal development of the lung. Annu Rev Physiol. 1984;46:617–628. - PubMed
-
- McGowan S.E. Paracrine cellular and extracellular matrix interactions with mesenchymal progenitors during pulmonary alveolar septation. Birth Defects Res A Clin Mol Teratol. 2014;100:227–239. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
