

Dietary and pharmacological modification of the insulin/IGF-1 system: exploiting the full repertoire against cancer
- PMID: 26878387
- PMCID: PMC5154349
- DOI: 10.1038/oncsis.2016.2
Dietary and pharmacological modification of the insulin/IGF-1 system: exploiting the full repertoire against cancer
Abstract
As more and more links between cancer and metabolism are discovered, new approaches to treat cancer using these mechanisms are considered. Dietary restriction of either calories or macronutrients has shown great potential in animal studies to both reduce the incidence and growth of cancer, and to act synergistically with other treatment strategies. These studies have also shown that dietary restriction simultaneously targets many of the molecular pathways that are targeted individually by anticancer drugs. The insulin/insulin-like growth factor-1 (IGF-1) system has thereby emerged as a key regulator of cancer growth pathways. Although lowering of insulin levels with diet or drugs such as metformin and diazoxide seems generally beneficial, some practitioners also utilize strategic elevations of insulin levels in combination with chemotherapeutic drugs. This indicates a broad spectrum of possibilities for modulating the insulin/IGF-1 system in cancer treatment. With a specific focus on dietary restriction, insulin administration and the insulin-lowering drug diazoxide, such modifications of the insulin/IGF-1 system are the topic of this review. Although preclinical data are promising, we point out that insulin regulation and the metabolic response to a certain diet often differ between mice and humans. Thus, the need for collecting more human data has to be emphasized.
Figures
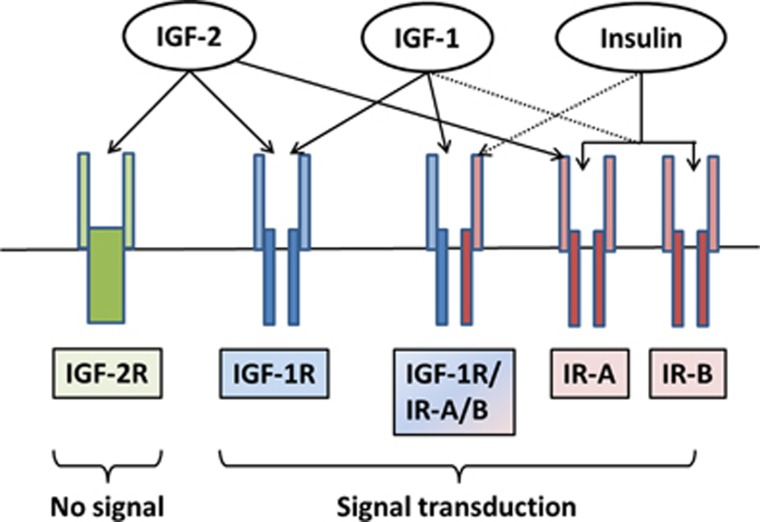
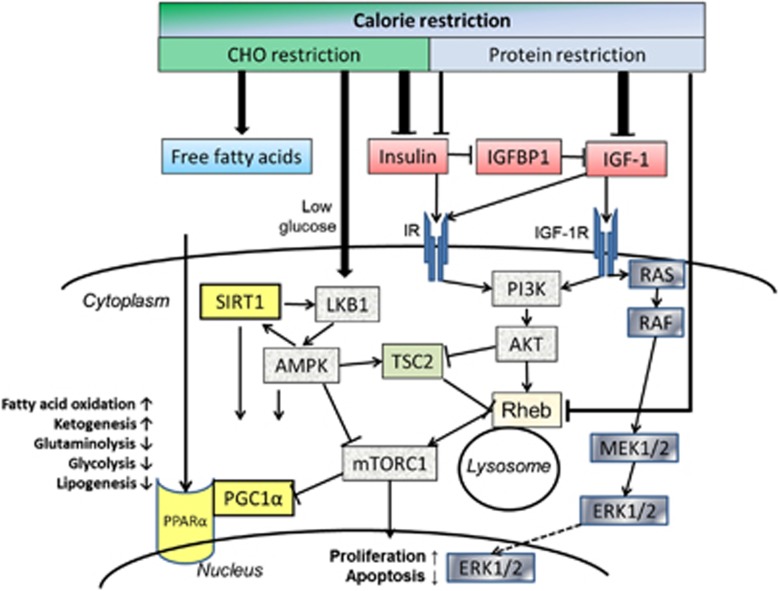
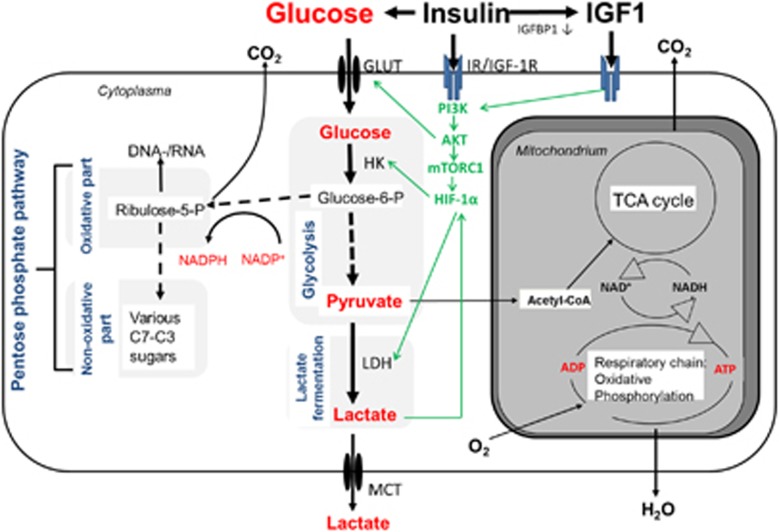
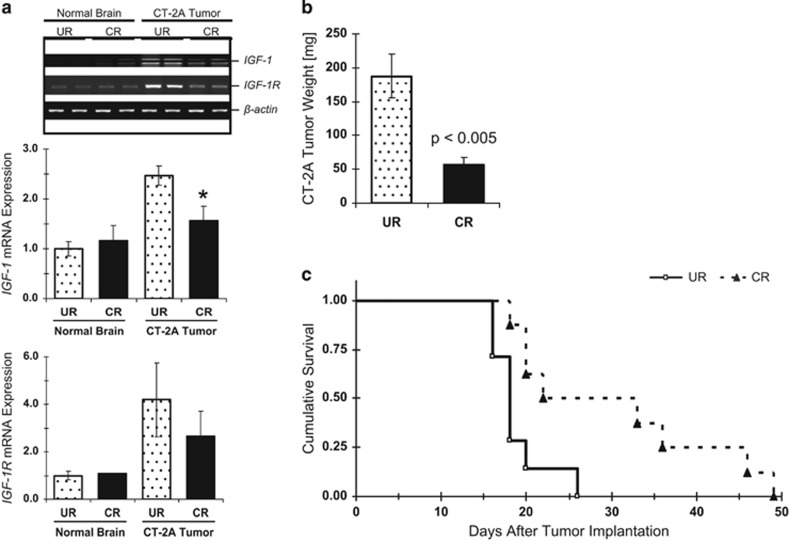
References
-
- Carrera-Bastos P, Fontes-Villalba M, ÓKeefe JH, Lindeberg S, Cordain L. The western diet and lifestyle and diseases of civilization. Res Rep Clin Cardiol 2011; 2: 15–35.
-
- Klement RJ, Gonder U, Orsó E, Paul S, Schilling F, Spitz J. Proceedings of the 2nd annual symposium of the German Society for Paleo Nutrition held in 2014. J Evo Health 2013; 1: 6.
-
- Ruiz-Núñez B, Pruimboom L, Dijck-Brouwer DAJ, Muskiet FAJ. Lifestyle and nutritional imbalances associated with Western diseases: causes and consequences of chronic systemic low-grade inflammation in an evolutionary context. J Nutr Biochem 2013; 24: 1183–1201. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
