

Killing Me Softly: Connotations to Unfolded Protein Response and Oxidative Stress in Alzheimer's Disease
- PMID: 26881014
- PMCID: PMC4736771
- DOI: 10.1155/2016/1805304
Killing Me Softly: Connotations to Unfolded Protein Response and Oxidative Stress in Alzheimer's Disease
Abstract
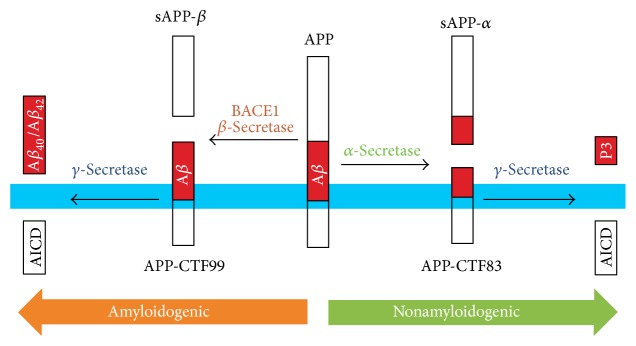
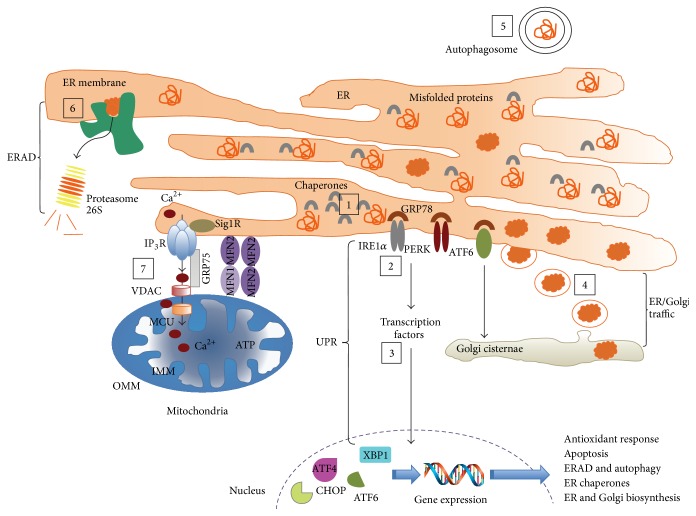
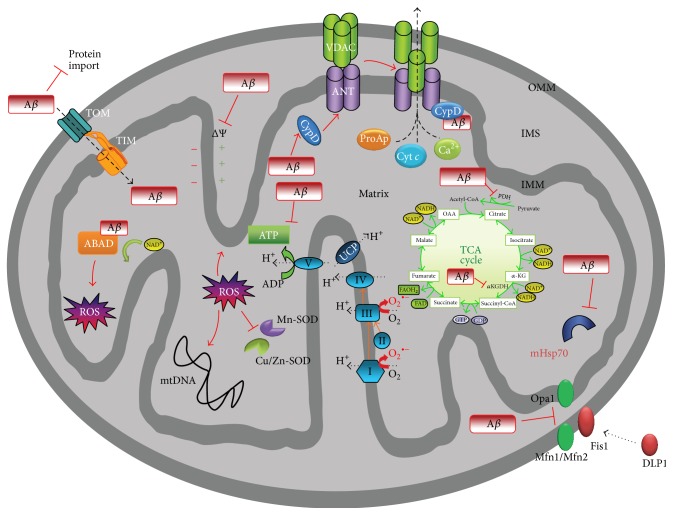
This review is focused on the possible causes of mitochondrial dysfunction in AD, underlying molecular mechanisms of this malfunction, possible causes and known consequences of APP, Aβ, and hyperphosphorylated tau presence in mitochondria, and the contribution of altered lipid metabolism (nonsterol isoprenoids) to pathological processes leading to increased formation and accumulation of the aforementioned hallmarks of AD. Abnormal protein folding and unfolded protein response seem to be the outcomes of impaired glycosylation due to metabolic disturbances in geranylgeraniol intermediary metabolism. The origin and consecutive fate of APP, Aβ, and tau are emphasized on intracellular trafficking apparently influenced by inaccurate posttranslational modifications. We hypothesize that incorrect intracellular processing of APP determines protein translocation to mitochondria in AD. Similarly, without obvious reasons, the passage of Aβ and tau to mitochondria is observed. APP targeted to mitochondria blocks the activity of protein translocase complex resulting in poor import of proteins central to oxidative phosphorylation. Besides, APP, Aβ, and neurofibrillary tangles of tau directly or indirectly impair mitochondrial biochemistry and bioenergetics, with concomitant generation of oxidative/nitrosative stress. Limited protective mechanisms are inadequate to prevent the free radical-mediated lesions. Finally, neuronal loss is observed in AD-affected brains typically by pathologic apoptosis.
Figures
Similar articles
-
Link between the unfolded protein response and dysregulation of mitochondrial bioenergetics in Alzheimer's disease.Cell Mol Life Sci. 2019 Apr;76(7):1419-1431. doi: 10.1007/s00018-019-03009-4. Epub 2019 Jan 25. Cell Mol Life Sci. 2019. PMID: 30683981 Free PMC article.
-
Mitochondria are a direct site of A beta accumulation in Alzheimer's disease neurons: implications for free radical generation and oxidative damage in disease progression.Hum Mol Genet. 2006 May 1;15(9):1437-49. doi: 10.1093/hmg/ddl066. Epub 2006 Mar 21. Hum Mol Genet. 2006. PMID: 16551656
-
Alzheimer's disease.Subcell Biochem. 2012;65:329-52. doi: 10.1007/978-94-007-5416-4_14. Subcell Biochem. 2012. PMID: 23225010 Review.
-
Impaired autophagy and APP processing in Alzheimer's disease: The potential role of Beclin 1 interactome.Prog Neurobiol. 2013 Jul-Aug;106-107:33-54. doi: 10.1016/j.pneurobio.2013.06.002. Epub 2013 Jul 1. Prog Neurobiol. 2013. PMID: 23827971 Review.
-
Mitochondria and vascular lesions as a central target for the development of Alzheimer's disease and Alzheimer disease-like pathology in transgenic mice.Neurol Res. 2003 Sep;25(6):665-74. doi: 10.1179/016164103101201977. Neurol Res. 2003. PMID: 14503022
Cited by
-
HSPA1A, HSPA2, and HSPA8 Are Potential Molecular Biomarkers for Prognosis among HSP70 Family in Alzheimer's Disease.Dis Markers. 2022 Sep 30;2022:9480398. doi: 10.1155/2022/9480398. eCollection 2022. Dis Markers. 2022. PMID: 36246562 Free PMC article.
-
A Mitocentric View of Alzheimer's Disease.Mol Neurobiol. 2017 Oct;54(8):6046-6060. doi: 10.1007/s12035-016-0117-7. Epub 2016 Oct 1. Mol Neurobiol. 2017. PMID: 27696116 Review.
-
Altered Nup153 Expression Impairs the Function of Cultured Hippocampal Neural Stem Cells Isolated from a Mouse Model of Alzheimer's Disease.Mol Neurobiol. 2019 Aug;56(8):5934-5949. doi: 10.1007/s12035-018-1466-1. Epub 2019 Jan 28. Mol Neurobiol. 2019. PMID: 30689197
-
Icariside II Ameliorates Cognitive Impairments Induced by Chronic Cerebral Hypoperfusion by Inhibiting the Amyloidogenic Pathway: Involvement of BDNF/TrkB/CREB Signaling and Up-Regulation of PPARα and PPARγ in Rats.Front Pharmacol. 2018 Oct 23;9:1211. doi: 10.3389/fphar.2018.01211. eCollection 2018. Front Pharmacol. 2018. PMID: 30405422 Free PMC article.
-
Emerging pathways driving early synaptic pathology in Alzheimer's disease.Biochem Biophys Res Commun. 2017 Feb 19;483(4):988-997. doi: 10.1016/j.bbrc.2016.09.088. Epub 2016 Sep 20. Biochem Biophys Res Commun. 2017. PMID: 27659710 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
