

Genetics and biology of pancreatic ductal adenocarcinoma
- PMID: 26883357
- PMCID: PMC4762423
- DOI: 10.1101/gad.275776.115
Genetics and biology of pancreatic ductal adenocarcinoma
Abstract
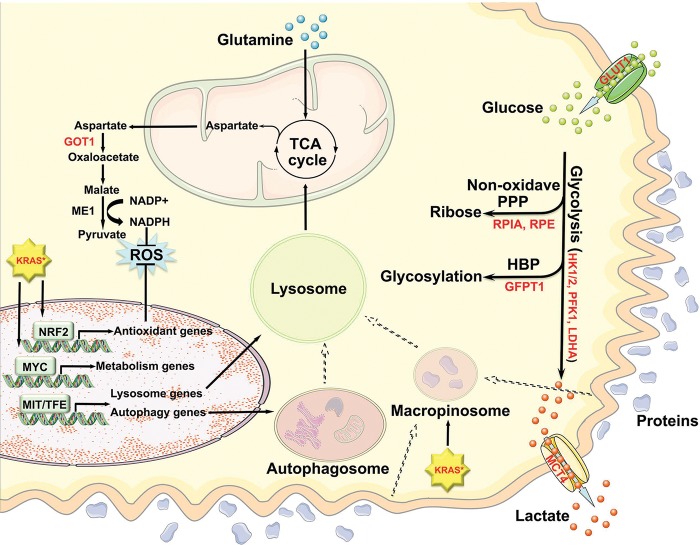
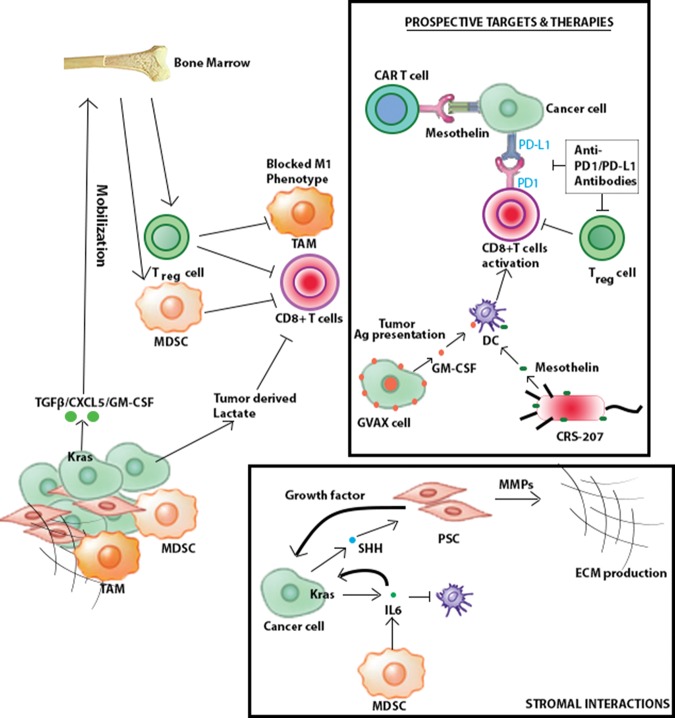
With 5-year survival rates remaining constant at 6% and rising incidences associated with an epidemic in obesity and metabolic syndrome, pancreatic ductal adenocarcinoma (PDAC) is on track to become the second most common cause of cancer-related deaths by 2030. The high mortality rate of PDAC stems primarily from the lack of early diagnosis and ineffective treatment for advanced tumors. During the past decade, the comprehensive atlas of genomic alterations, the prominence of specific pathways, the preclinical validation of such emerging targets, sophisticated preclinical model systems, and the molecular classification of PDAC into specific disease subtypes have all converged to illuminate drug discovery programs with clearer clinical path hypotheses. A deeper understanding of cancer cell biology, particularly altered cancer cell metabolism and impaired DNA repair processes, is providing novel therapeutic strategies that show strong preclinical activity. Elucidation of tumor biology principles, most notably a deeper understanding of the complexity of immune regulation in the tumor microenvironment, has provided an exciting framework to reawaken the immune system to attack PDAC cancer cells. While the long road of translation lies ahead, the path to meaningful clinical progress has never been clearer to improve PDAC patient survival.
Keywords: KRAS; cancer metabolism; pancreatic cancer; tumor immune modulation.
© 2016 Ying et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
Comment in
-
Upregulation of brain cholesterol levels inhibits mitophagy in Alzheimer disease.Autophagy. 2021 Jun;17(6):1555-1557. doi: 10.1080/15548627.2021.1920814. Epub 2021 May 4. Autophagy. 2021. PMID: 33945386 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous