

A MED13-dependent skeletal muscle gene program controls systemic glucose homeostasis and hepatic metabolism
- PMID: 26883362
- PMCID: PMC4762428
- DOI: 10.1101/gad.273128.115
A MED13-dependent skeletal muscle gene program controls systemic glucose homeostasis and hepatic metabolism
Abstract
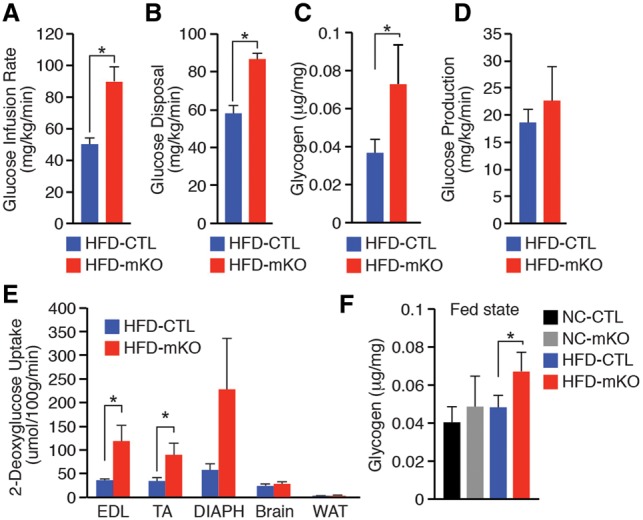
The Mediator complex governs gene expression by linking upstream signaling pathways with the basal transcriptional machinery. However, how individual Mediator subunits may function in different tissues remains to be investigated. Through skeletal muscle-specific deletion of the Mediator subunit MED13 in mice, we discovered a gene regulatory mechanism by which skeletal muscle modulates the response of the liver to a high-fat diet. Skeletal muscle-specific deletion of MED13 in mice conferred resistance to hepatic steatosis by activating a metabolic gene program that enhances muscle glucose uptake and storage as glycogen. The consequent insulin-sensitizing effect within skeletal muscle lowered systemic glucose and insulin levels independently of weight gain and adiposity and prevented hepatic lipid accumulation. MED13 suppressed the expression of genes involved in glucose uptake and metabolism in skeletal muscle by inhibiting the nuclear receptor NURR1 and the MEF2 transcription factor. These findings reveal a fundamental molecular mechanism for the governance of glucose metabolism and the control of hepatic lipid accumulation by skeletal muscle. Intriguingly, MED13 exerts opposing metabolic actions in skeletal muscle and the heart, highlighting the customized, tissue-specific functions of the Mediator complex.
Keywords: NURR1/NR4A2; glucose homeostasis; mediator complex; skeletal muscle.
© 2016 Amoasii et al.; Published by Cold Spring Harbor Laboratory Press.
Figures





References
-
- Ayala JE, Bracy DP, McGuinness OP, Wasserman DH. 2006. Considerations in the design of hyperinsulinemic-euglycemic clamps in the conscious mouse. Diabetes 55: 390–397. - PubMed
-
- Berdeaux R, Goebel N, Banaszynski L, Takemori H, Wandless T, Shelton GD, Montminy M. 2007. SIK1 is a class II HDAC kinase that promotes survival of skeletal myocytes. Nat Med 13: 597–603. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- K99 DK094973/DK/NIDDK NIH HHS/United States
- R01 HL077439/HL/NHLBI NIH HHS/United States
- R01 DK099653/DK/NIDDK NIH HHS/United States
- DK-099653/DK/NIDDK NIH HHS/United States
- DK-094973/DK/NIDDK NIH HHS/United States
- U01 HL100401/HL/NHLBI NIH HHS/United States
- R00 DK094973/DK/NIDDK NIH HHS/United States
- R01 HL111665/HL/NHLBI NIH HHS/United States
- HL-077439/HL/NHLBI NIH HHS/United States
- U01-HL-100401/HL/NHLBI NIH HHS/United States
- HL-111665/HL/NHLBI NIH HHS/United States
- R01 AR067294/AR/NIAMS NIH HHS/United States
- R01 HL093039/HL/NHLBI NIH HHS/United States
- HL093039/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases