

Cardiac-Specific Deletion of the Pdha1 Gene Sensitizes Heart to Toxicological Actions of Ischemic Stress
- PMID: 26884059
- PMCID: PMC4914805
- DOI: 10.1093/toxsci/kfw035
Cardiac-Specific Deletion of the Pdha1 Gene Sensitizes Heart to Toxicological Actions of Ischemic Stress
Erratum in
-
Cardiac-Specific Deletion of the Pdha1 Gene Sensitizes Heart to Toxicological Actions of Ischemic Stress.Toxicol Sci. 2016 Oct;153(2):411. doi: 10.1093/toxsci/kfw154. Epub 2016 Sep 7. Toxicol Sci. 2016. PMID: 27605416 Free PMC article. No abstract available.
Abstract
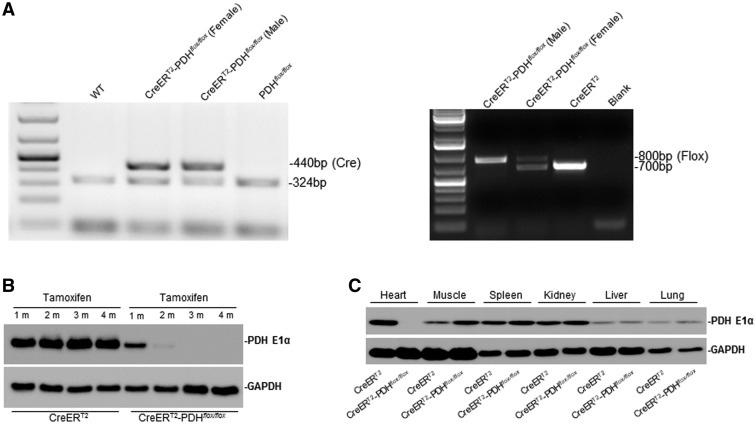
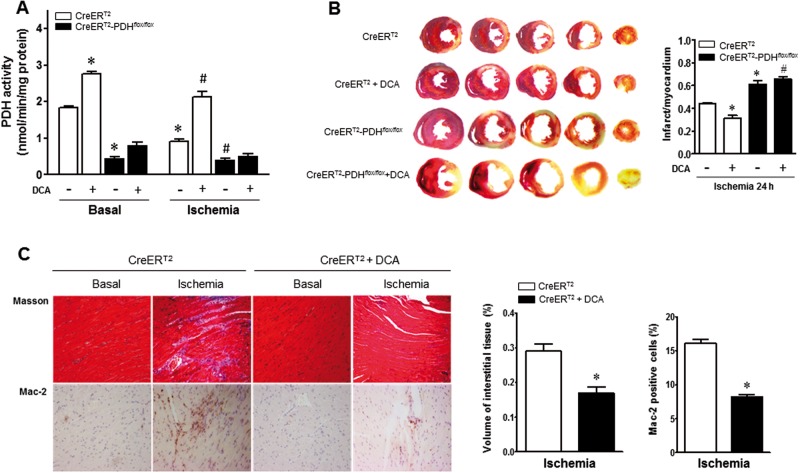
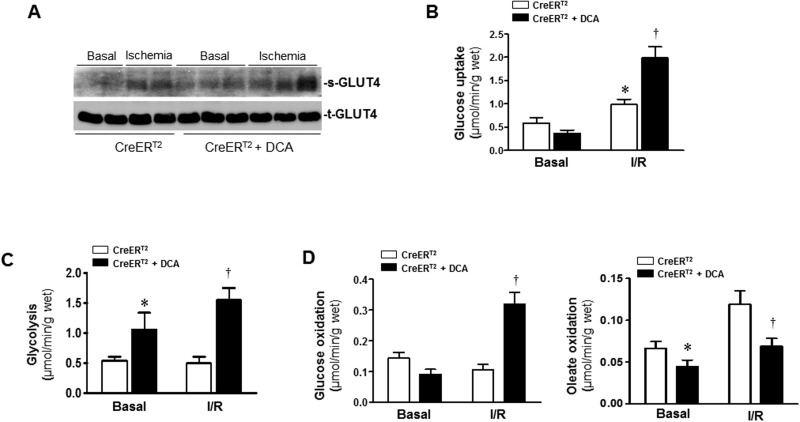
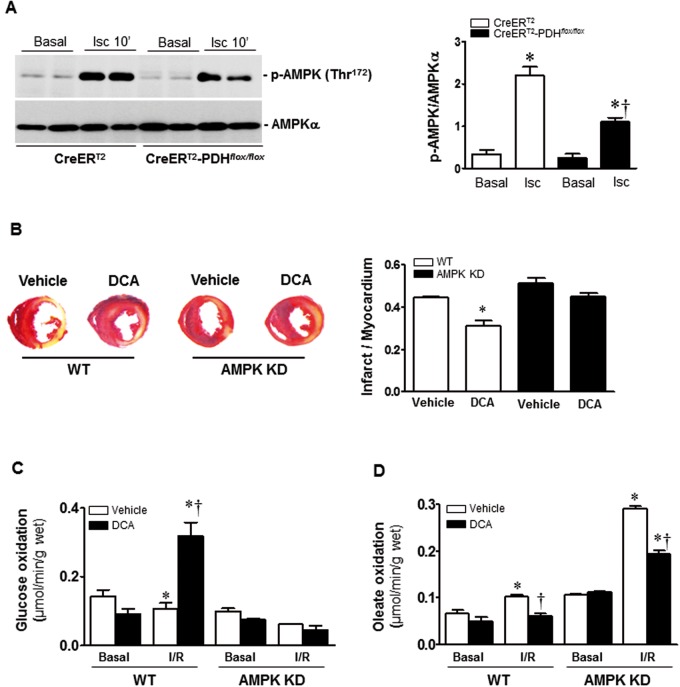
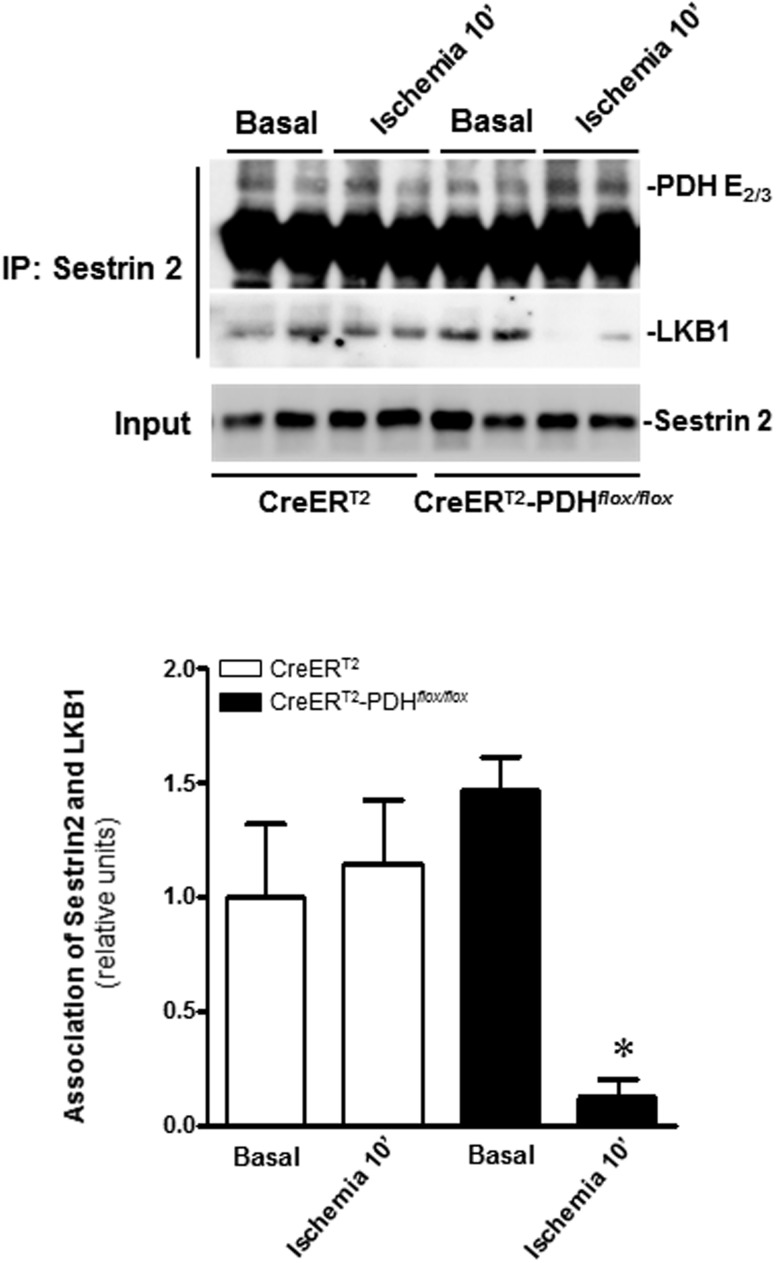
Pyruvate dehydrogenase (PDH) plays a key role in aerobic energy metabolism and occupies a central crossroad between glycolysis and the tricarboxylic acid cycle. We generated inducible cardiac-specific PDH E1α knockout (CreER(T2)-PDH(flox/flox)) mice that demonstrated a high mortality rate. It was hypothesized that PDH modulating cardiac glucose metabolism is crucial for heart functions under normal physiological and/or stress conditions. The myocardial infarction was conducted by a ligation of the left anterior descending coronary arteries. Cardiac PDH E1α deficiency caused large myocardial infarcts size and macrophage infiltration in the hearts (P < .01 vs wild-type [WT]). Wheat germ agglutinin and Masson trichrome staining revealed significantly increased hypertrophy and fibrosis in PDH E1α-deficient hearts (P < .05 vs WT). Measurements of heart substrate metabolism in an ex vivo working heart perfusion system demonstrated a significant impairment of glucose oxidation in PDH E1α-deficient hearts during ischemia/reperfusion (P < .05 vs WT). Dichloroacetate, a PDH activator, increased glucose oxidation in WT hearts during ischemia/reperfusion and reduced myocardial infarct size in WT, but not in PDH E1α-deficient hearts. Immunoblotting results demonstrated that cardiac PDH E1α deficiency leads to an impaired ischemic AMP-activated protein kinase activation through Sestrin2-liver kinase B1 interaction which is responsible for an increased susceptibility of PDH E1α-deficient heart to ischemic insults. Thus, cardiac PDH E1α deficiency impairs ischemic AMP-activated protein kinase signaling and sensitizes hearts to the toxicological actions of ischemic stress.
Keywords: AMP-activated protein kinase.; myocardial infarction; pyruvate dehydrogenase.
© The Author 2016. Published by Oxford University Press on behalf of the Society of Toxicology. All rights reserved. For Permissions, please e-mail: journals.permissions@oup.com.
Figures

References
-
- Anderson J., Khou S., Nawarskas J. (2000). Ranolazine: A potential new treatment for chronic stable angina. Heart Dis. 3, 263–269. - PubMed
-
- Bainey K. R., Armstrong P. W. (2014). Clinical perspectives on reperfusion injury in acute myocardial infarction. Am. Heart J. 167, 637–645. - PubMed
-
- Bersin R. M., Stacpoole P. W. (1997). Dichloroacetate as metabolic therapy for myocardial ischemia and failure. Am. Heart J. 134, 841–855. - PubMed
-
- Calvert J. W., Gundewar S., Jha S., Greer J. J., Bestermann W. H., Tian R., Lefer D. J. (2008). Acute metformin therapy confers cardioprotection against myocardial infarction via AMPK-eNOS-mediated signaling. Diabetes 57, 696–705. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
