

Deep phenotyping of 89 xeroderma pigmentosum patients reveals unexpected heterogeneity dependent on the precise molecular defect
- PMID: 26884178
- PMCID: PMC4780618
- DOI: 10.1073/pnas.1519444113
Deep phenotyping of 89 xeroderma pigmentosum patients reveals unexpected heterogeneity dependent on the precise molecular defect
Abstract
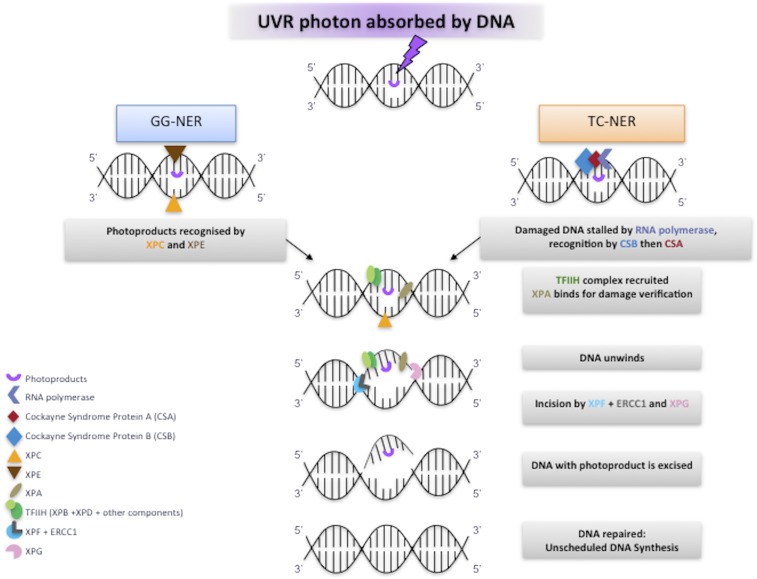
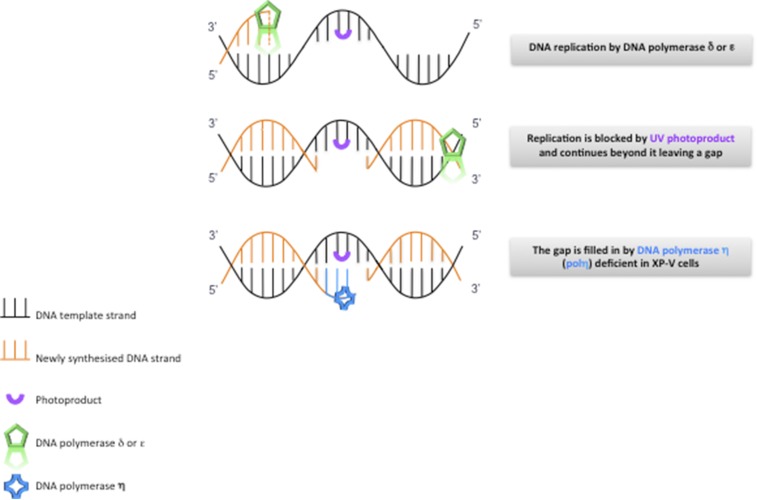
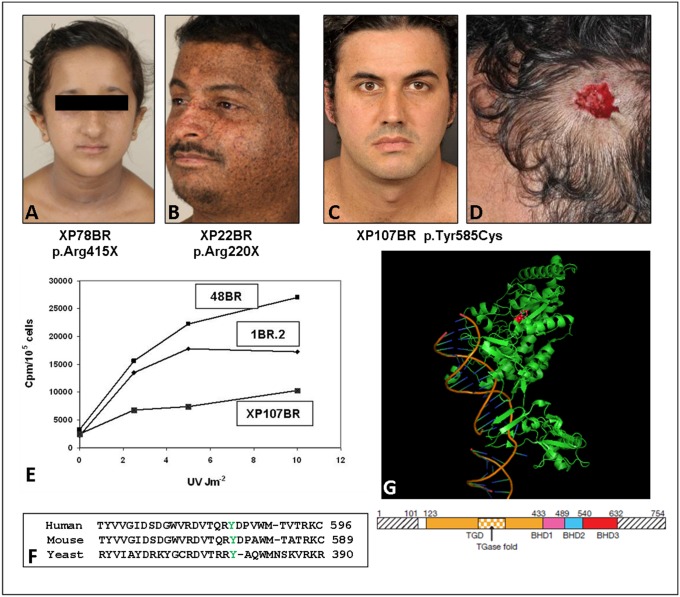
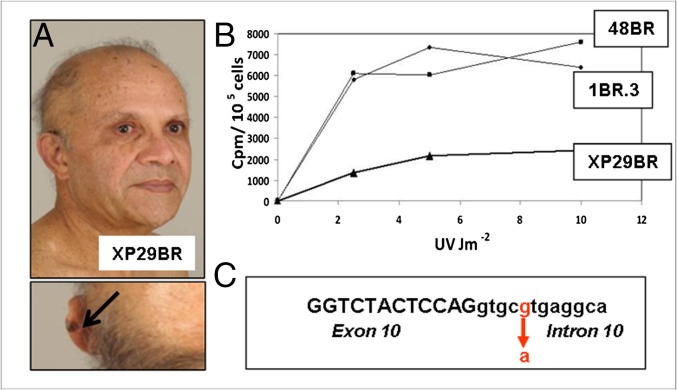
Xeroderma pigmentosum (XP) is a rare DNA repair disorder characterized by increased susceptibility to UV radiation (UVR)-induced skin pigmentation, skin cancers, ocular surface disease, and, in some patients, sunburn and neurological degeneration. Genetically, it is assigned to eight complementation groups (XP-A to -G and variant). For the last 5 y, the UK national multidisciplinary XP service has provided follow-up for 89 XP patients, representing most of the XP patients in the United Kingdom. Causative mutations, DNA repair levels, and more than 60 clinical variables relating to dermatology, ophthalmology, and neurology have been measured, using scoring systems to categorize disease severity. This deep phenotyping has revealed unanticipated heterogeneity of clinical features, between and within complementation groups. Skin cancer is most common in XP-C, XP-E, and XP-V patients, previously considered to be the milder groups based on cellular analyses. These patients have normal sunburn reactions and are therefore diagnosed later and are less likely to adhere to UVR protection. XP-C patients are specifically hypersensitive to ocular damage, and XP-F and XP-G patients appear to be much less susceptible to skin cancer than other XP groups. Within XP groups, different mutations confer susceptibility or resistance to neurological damage. Our findings on this large cohort of XP patients under long-term follow-up reveal that XP is more heterogeneous than has previously been appreciated. Our data now enable provision of personalized prognostic information and management advice for each XP patient, as well as providing new insights into the functions of the XP proteins.
Keywords: UV radiation; neurodegeneration; nucleotide excision repair; ocular disease; skin cancer.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
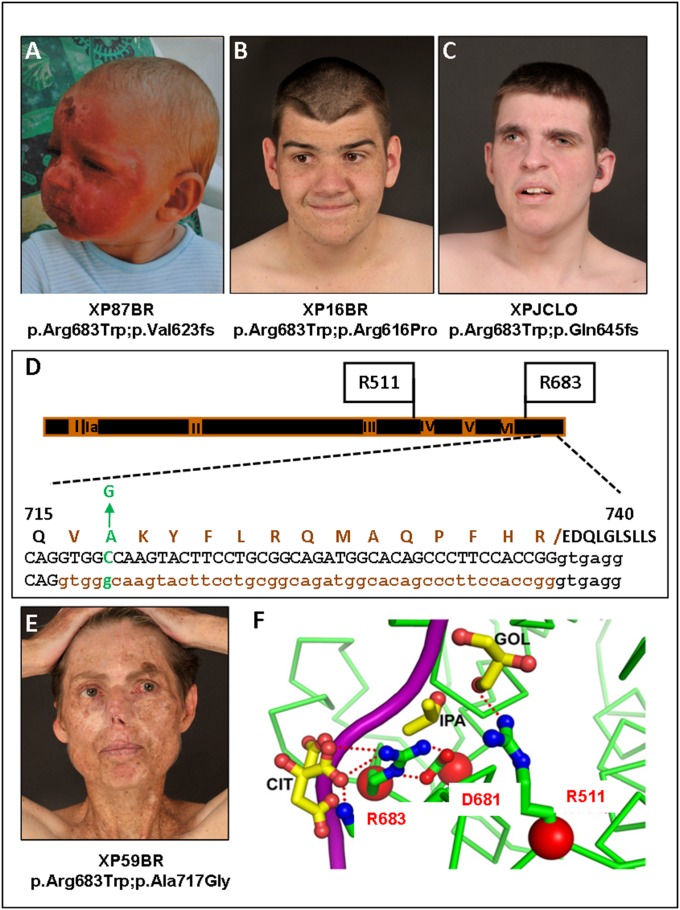
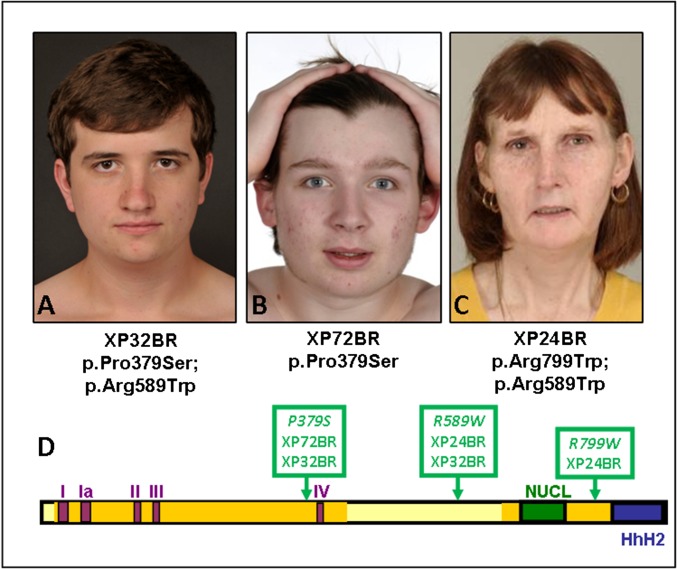
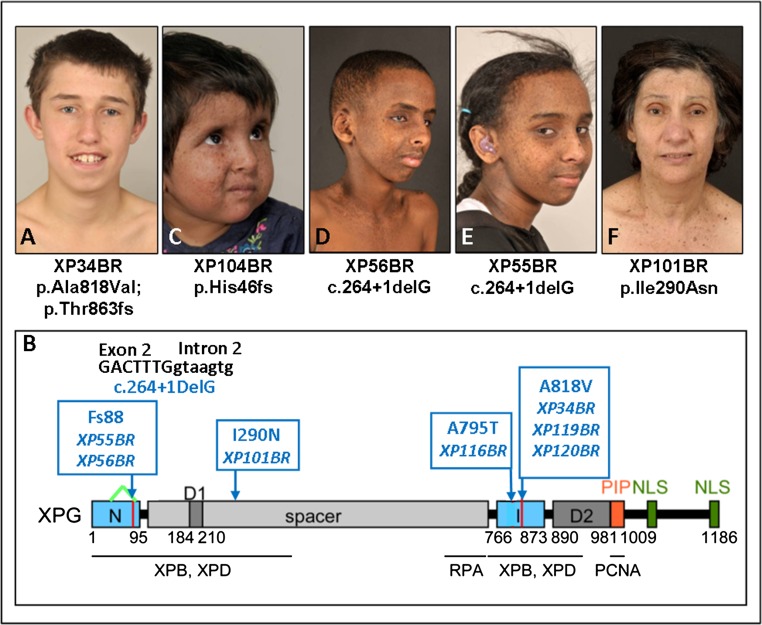
References
-
- Kleijer WJ, et al. Incidence of DNA repair deficiency disorders in western Europe: Xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. DNA Repair (Amst) 2008;7(5):744–750. - PubMed
-
- Hirai Y, et al. Heterozygous individuals bearing a founder mutation in the XPA DNA repair gene comprise nearly 1% of the Japanese population. Mutat Res. 2006;601(1-2):171–178. - PubMed
-
- Zghal M, et al. Xeroderma pigmentosum: Manifestations cutanées, oculaires et neurologiques à partir de 49 patients tunisiens. Tunis Med. 2005;83(12):760–763. - PubMed
-
- Sethi M, et al. Patients with xeroderma pigmentosum complementation groups C, E and V do not have abnormal sunburn reactions. Br J Dermatol. 2013;169(6):1279–1287. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
