

First structure of full-length mammalian phenylalanine hydroxylase reveals the architecture of an autoinhibited tetramer
- PMID: 26884182
- PMCID: PMC4780608
- DOI: 10.1073/pnas.1516967113
First structure of full-length mammalian phenylalanine hydroxylase reveals the architecture of an autoinhibited tetramer
Abstract
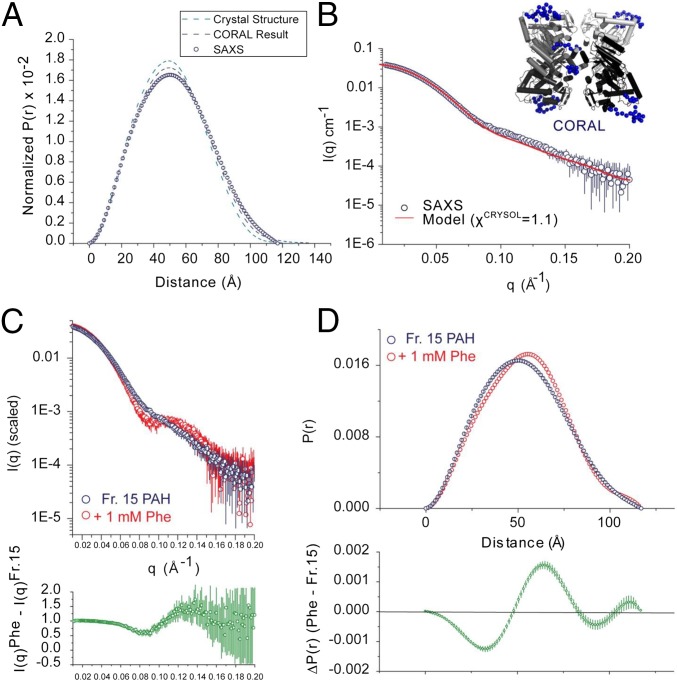
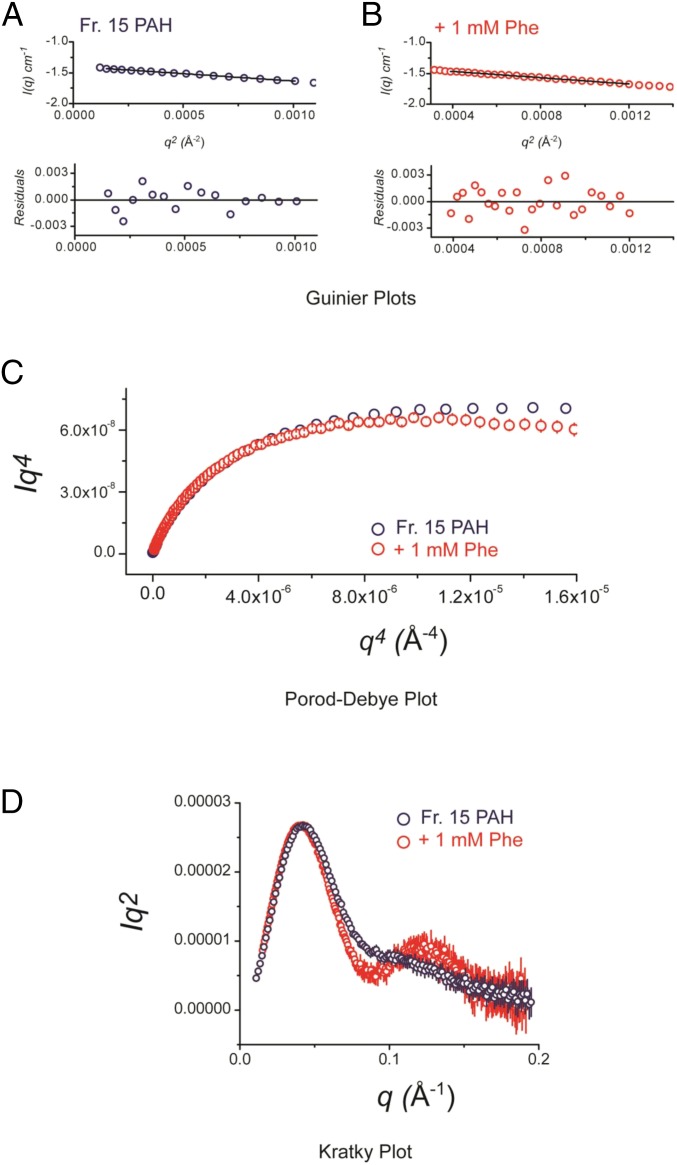
Improved understanding of the relationship among structure, dynamics, and function for the enzyme phenylalanine hydroxylase (PAH) can lead to needed new therapies for phenylketonuria, the most common inborn error of amino acid metabolism. PAH is a multidomain homo-multimeric protein whose conformation and multimerization properties respond to allosteric activation by the substrate phenylalanine (Phe); the allosteric regulation is necessary to maintain Phe below neurotoxic levels. A recently introduced model for allosteric regulation of PAH involves major domain motions and architecturally distinct PAH tetramers [Jaffe EK, Stith L, Lawrence SH, Andrake M, Dunbrack RL, Jr (2013) Arch Biochem Biophys 530(2):73-82]. Herein, we present, to our knowledge, the first X-ray crystal structure for a full-length mammalian (rat) PAH in an autoinhibited conformation. Chromatographic isolation of a monodisperse tetrameric PAH, in the absence of Phe, facilitated determination of the 2.9 Å crystal structure. The structure of full-length PAH supersedes a composite homology model that had been used extensively to rationalize phenylketonuria genotype-phenotype relationships. Small-angle X-ray scattering (SAXS) confirms that this tetramer, which dominates in the absence of Phe, is different from a Phe-stabilized allosterically activated PAH tetramer. The lack of structural detail for activated PAH remains a barrier to complete understanding of phenylketonuria genotype-phenotype relationships. Nevertheless, the use of SAXS and X-ray crystallography together to inspect PAH structure provides, to our knowledge, the first complete view of the enzyme in a tetrameric form that was not possible with prior partial crystal structures, and facilitates interpretation of a wealth of biochemical and structural data that was hitherto impossible to evaluate.
Keywords: X-ray crystallography; allosteric regulation; phenylalanine hydroxylase; phenylketonuria; small-angle X-ray scattering.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
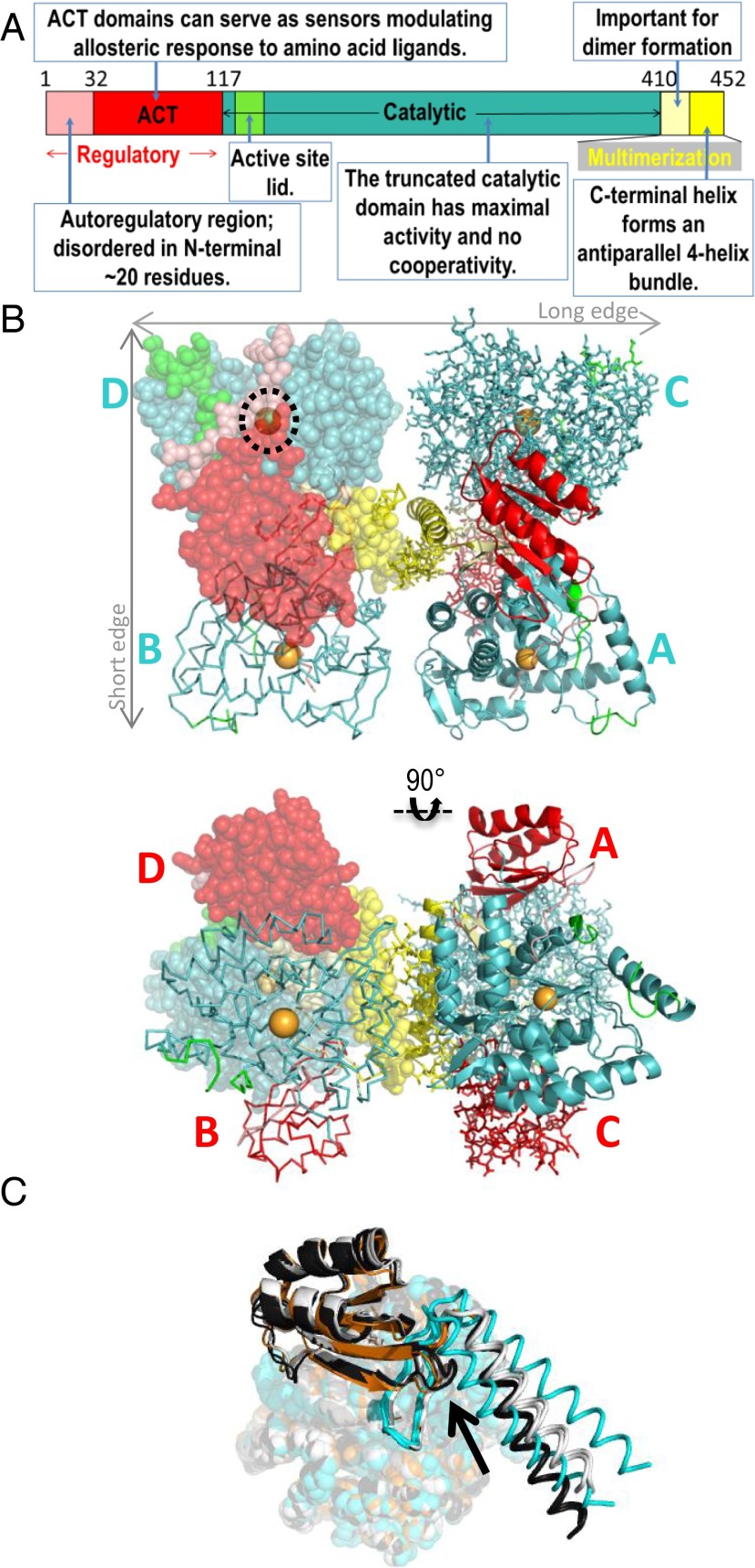
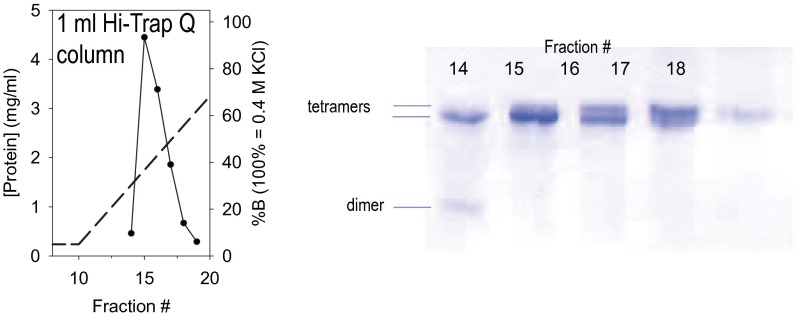
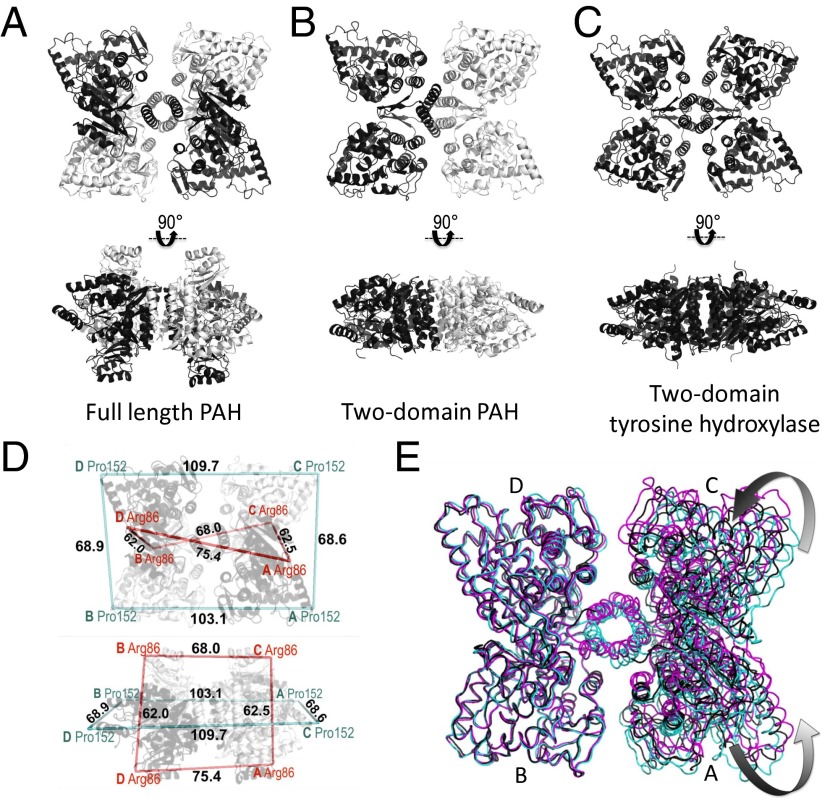
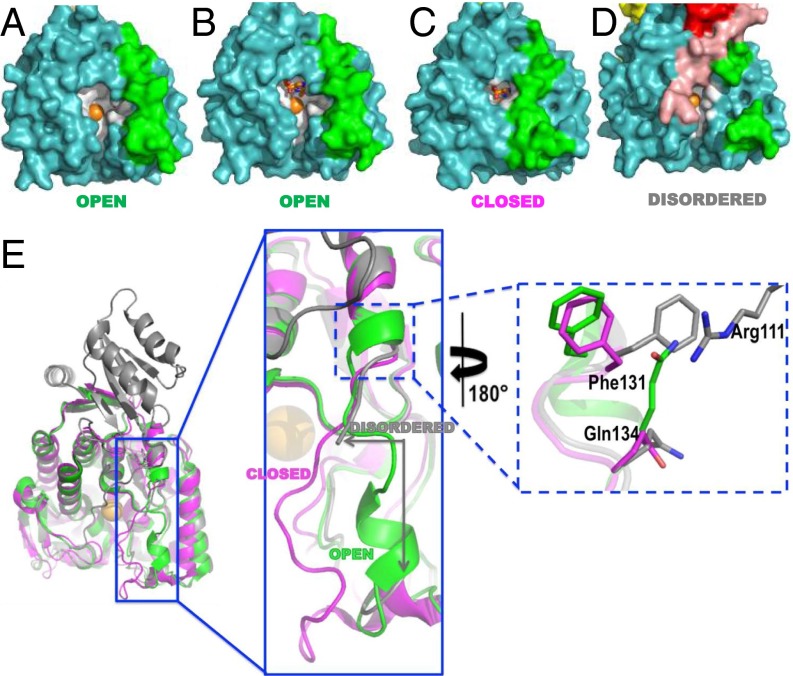
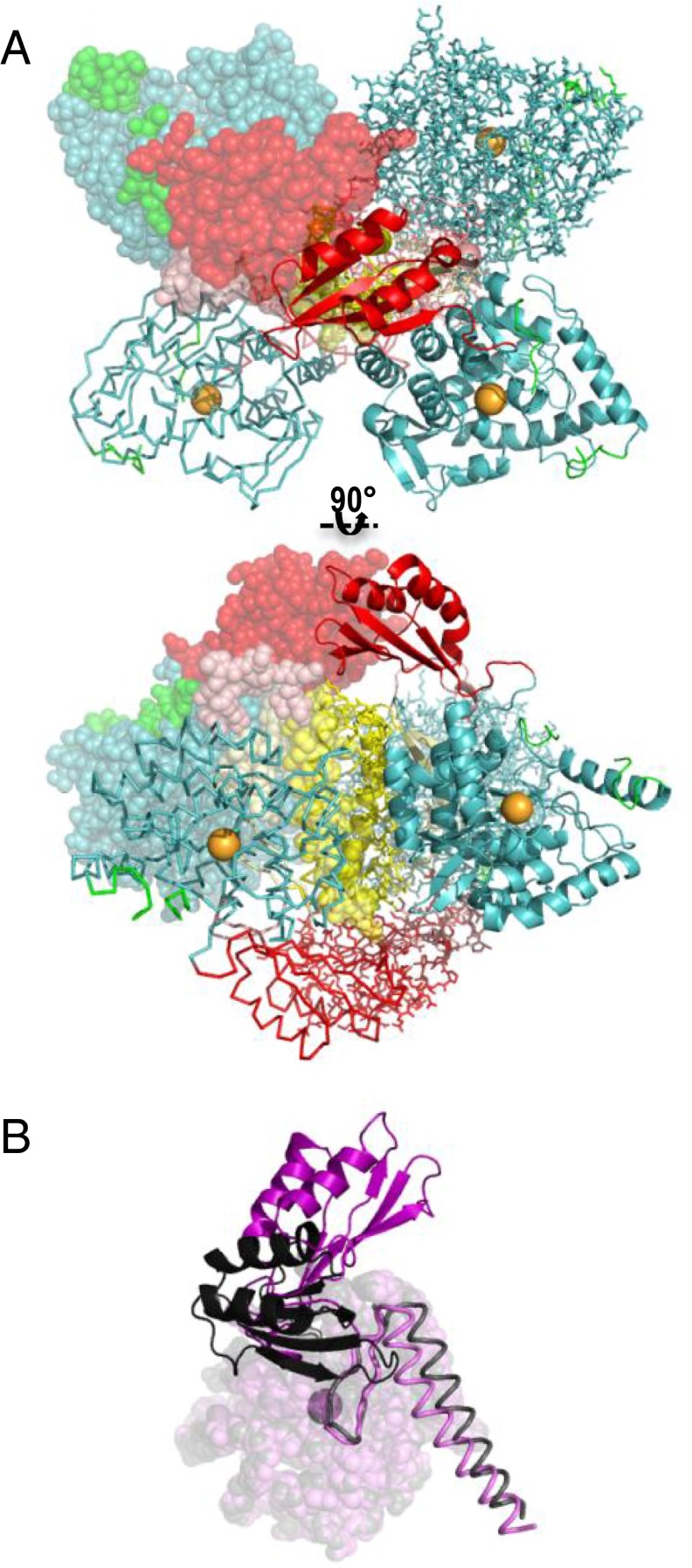
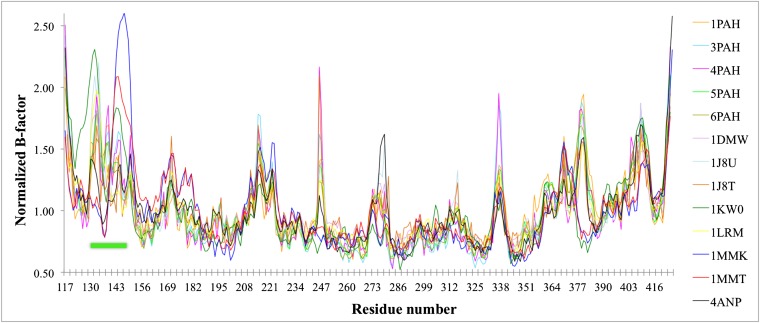
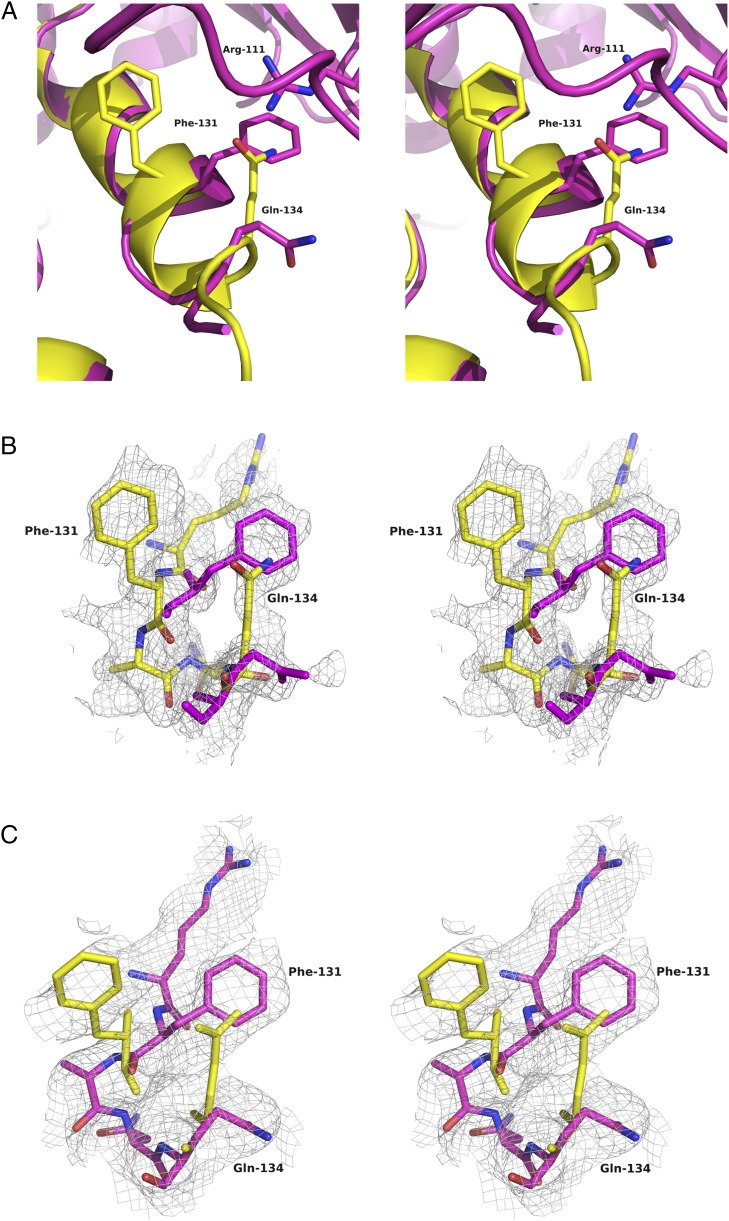
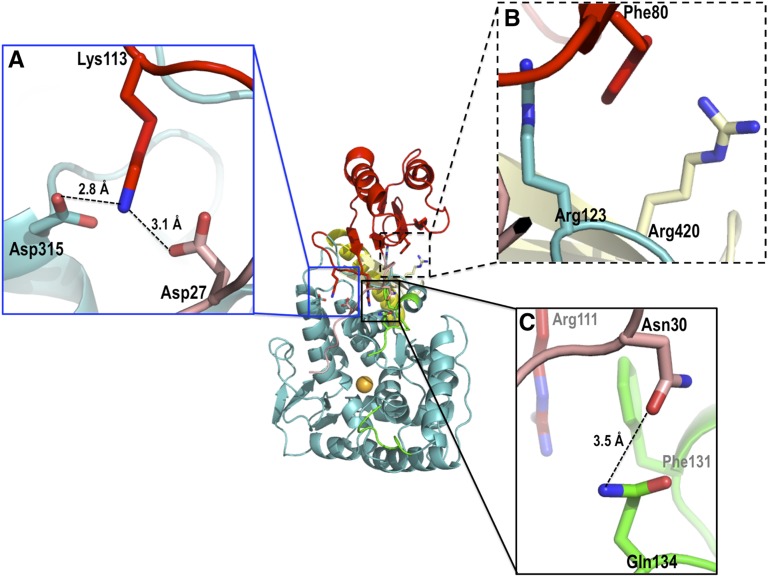
References
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
