

Smads as therapeutic targets for chronic kidney disease
- PMID: 26889404
- PMCID: PMC4715089
- DOI: 10.1016/j.krcp.2011.12.001
Smads as therapeutic targets for chronic kidney disease
Abstract
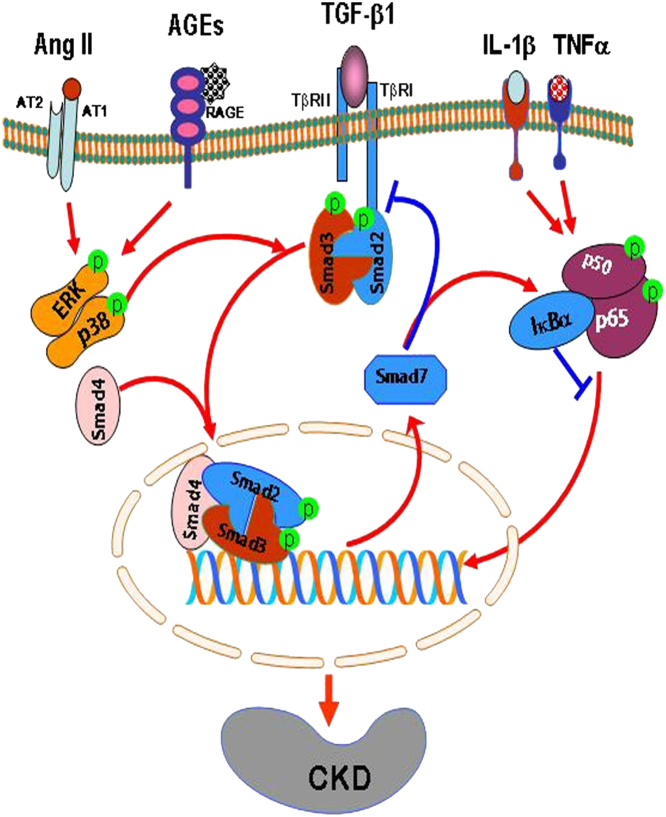
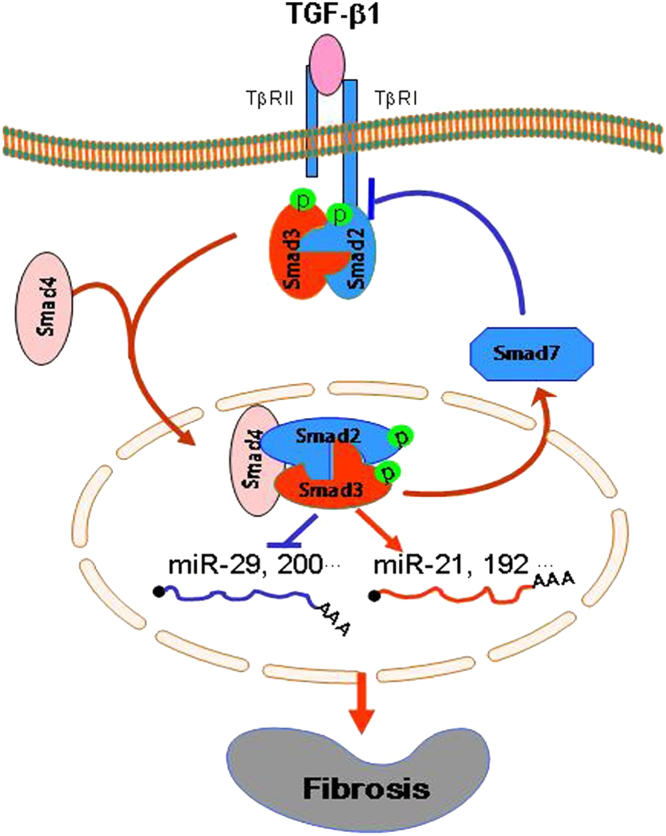
Renal fibrosis is a hallmark of chronic kidney disease (CKD). It is generally thought that transforming growth factor-β1 (TGF-β1) is a key mediator of fibrosis and mediates renal scarring positively by Smad2 and Smad3, but negatively by Smad7. Our recent studies found that in CKD, TGF-β1 is not a sole molecule to activate Smads. Many mediators such as angiotensin II and advanced glycation end products can also activate Smads via both TGF-β-dependent and independent mechanisms. In addition, Smads can interact with other signaling pathways, such as the mitogen-activated protein kinase and nuclear factor-kappaB (NF-κB) pathways, to regulate renal inflammation and fibrosis. In CKD, Smad2 and Smad3 are highly activated, while Smad7 is reduced or lost. In the context of fibrosis, Smad3 is pathogenic and mediates renal fibrosis by upregulating miR-21 and miR-192, but down-regulating miR-29 and miR-200 families. By contrast, Smad2 and Smad7 are protective. Overexpression of Smad7 inhibits both Smad3-mediated renal fibrosis and NF-κB-driven renal inflammation. Interestingly, Smad4 has diverse roles in renal fibrosis and inflammation. The complexity and distinct roles of individual Smads in CKD suggest that treatment of CKD should aim to correct the imbalance of Smad signaling or target the Smad3-dependent genes related to fibrosis, rather than to block the general effect of TGF-β1. Thus, treatment of CKD by overexpression of Smad7 or targeting Smad3-dependent miRNAs such as downregulation of miR-21 or overexpression of miR-29 may represent novel therapeutic strategies for CKD.
Keywords: Chronic kidney disease; Fibrosis; Gene therapy; Inflammation; MicroRNA; TGF-β/Smads.
Figures
Similar articles
-
Diverse roles of TGF-β/Smads in renal fibrosis and inflammation.Int J Biol Sci. 2011;7(7):1056-67. doi: 10.7150/ijbs.7.1056. Epub 2011 Sep 2. Int J Biol Sci. 2011. PMID: 21927575 Free PMC article. Review.
-
Central role of dysregulation of TGF-β/Smad in CKD progression and potential targets of its treatment.Biomed Pharmacother. 2018 May;101:670-681. doi: 10.1016/j.biopha.2018.02.090. Epub 2018 Mar 22. Biomed Pharmacother. 2018. PMID: 29518614 Review.
-
TGF-β/Smad signaling in renal fibrosis.Front Physiol. 2015 Mar 19;6:82. doi: 10.3389/fphys.2015.00082. eCollection 2015. Front Physiol. 2015. PMID: 25852569 Free PMC article. Review.
-
Transforming growth factor-β/Smad signalling in diabetic nephropathy.Clin Exp Pharmacol Physiol. 2012 Aug;39(8):731-8. doi: 10.1111/j.1440-1681.2011.05663.x. Clin Exp Pharmacol Physiol. 2012. PMID: 22211842 Review.
-
TGF-β/Smad signaling in kidney disease.Semin Nephrol. 2012 May;32(3):236-43. doi: 10.1016/j.semnephrol.2012.04.002. Semin Nephrol. 2012. PMID: 22835454 Review.
Cited by
-
β-Mangostin Alleviates Renal Tubulointerstitial Fibrosis via the TGF-β1/JNK Signaling Pathway.Cells. 2024 Oct 14;13(20):1701. doi: 10.3390/cells13201701. Cells. 2024. PMID: 39451219 Free PMC article.
-
Circulating extracellular vesicles in human cardiorenal syndrome promote renal injury in a kidney-on-chip system.JCI Insight. 2023 Nov 22;8(22):e165172. doi: 10.1172/jci.insight.165172. JCI Insight. 2023. PMID: 37707956 Free PMC article.
-
Retinoic acid receptor α activity in proximal tubules prevents kidney injury and fibrosis.Proc Natl Acad Sci U S A. 2024 Feb 13;121(7):e2311803121. doi: 10.1073/pnas.2311803121. Epub 2024 Feb 8. Proc Natl Acad Sci U S A. 2024. PMID: 38330015 Free PMC article.
-
Effects of tranilast on the epithelial-to-mesenchymal transition in peritoneal mesothelial cells.Kidney Res Clin Pract. 2019 Dec 31;38(4):472-480. doi: 10.23876/j.krcp.19.049. Kidney Res Clin Pract. 2019. PMID: 31554027 Free PMC article.
-
HL156A, a novel pharmacological agent with potent adenosine-monophosphate-activated protein kinase (AMPK) activator activity ameliorates renal fibrosis in a rat unilateral ureteral obstruction model.PLoS One. 2018 Aug 30;13(8):e0201692. doi: 10.1371/journal.pone.0201692. eCollection 2018. PLoS One. 2018. PMID: 30161162 Free PMC article.
References
-
- Eddy A.A., Neilson E.G. Chronic kidney disease progression. J Am Soc Nephrol. 2006;17:2964–2966. - PubMed
-
- Bottinger E.P. TGF-beta in renal injury and disease. Semin Nephrol. 2007;27:309–320. - PubMed
-
- Wang W., Koka V., Lan H.Y. Transforming growth factor-beta and Smad signalling in kidney diseases. Nephrology (Carlton) 2005;10:43–56. - PubMed
-
- Roberts A.B. Molecular and cell biology of TGF-beta. Miner Electrolyte Metab. 1998;24:111–119. - PubMed
-
- Derynck R., Zhang Y.E. Smad-dependent and Smad-independent pathways in TGFbeta family signalling. Nature. 2003;425:577–584. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
