

Establishing the precise evolutionary history of a gene improves prediction of disease-causing missense mutations
- PMID: 26890452
- PMCID: PMC4990510
- DOI: 10.1038/gim.2015.208
Establishing the precise evolutionary history of a gene improves prediction of disease-causing missense mutations
Abstract
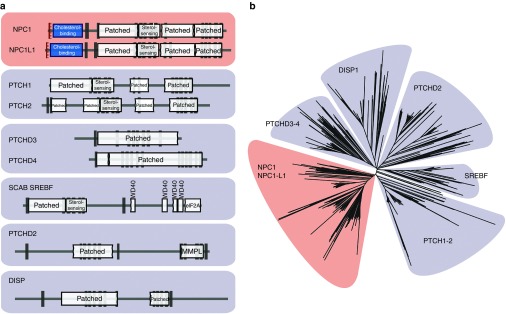
Purpose: Predicting the phenotypic effects of mutations has become an important application in clinical genetic diagnostics. Computational tools evaluate the behavior of the variant over evolutionary time and assume that variations seen during the course of evolution are probably benign in humans. However, current tools do not take into account orthologous/paralogous relationships. Paralogs have dramatically different roles in Mendelian diseases. For example, whereas inactivating mutations in the NPC1 gene cause the neurodegenerative disorder Niemann-Pick C, inactivating mutations in its paralog NPC1L1 are not disease-causing and, moreover, are implicated in protection from coronary heart disease.
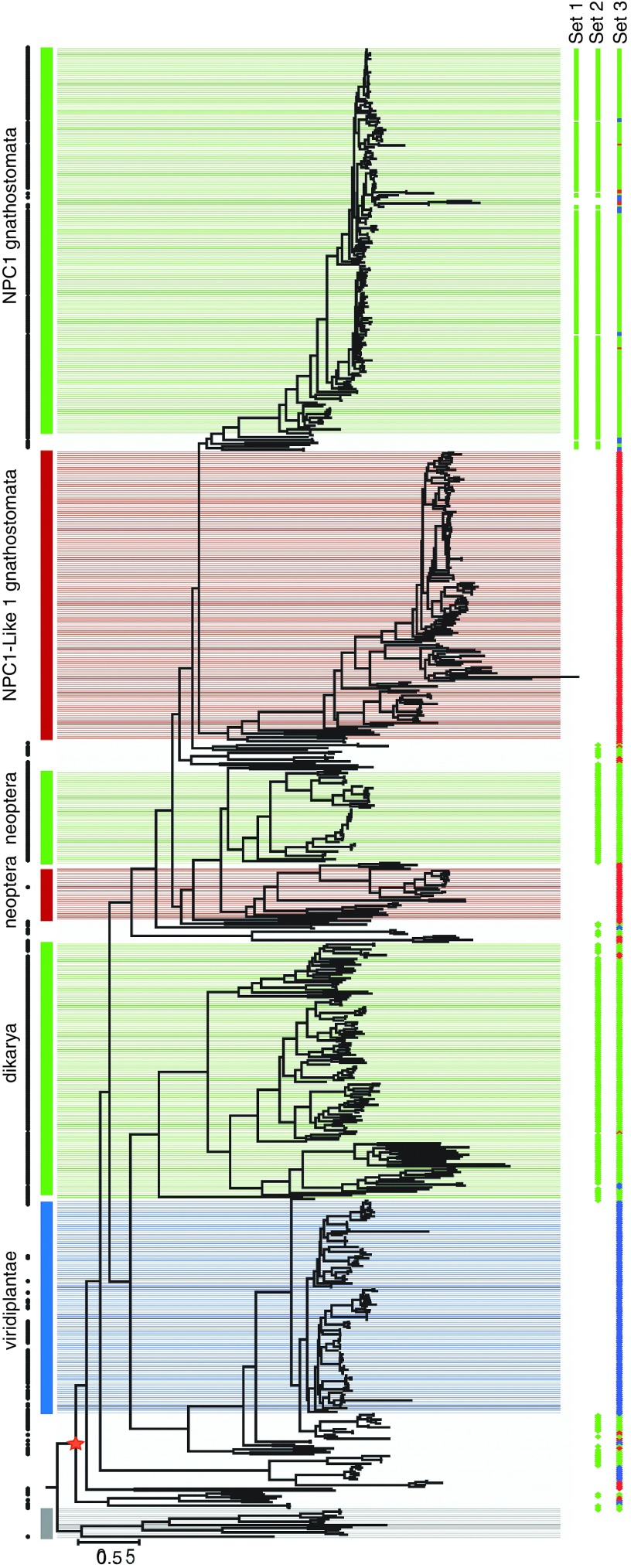
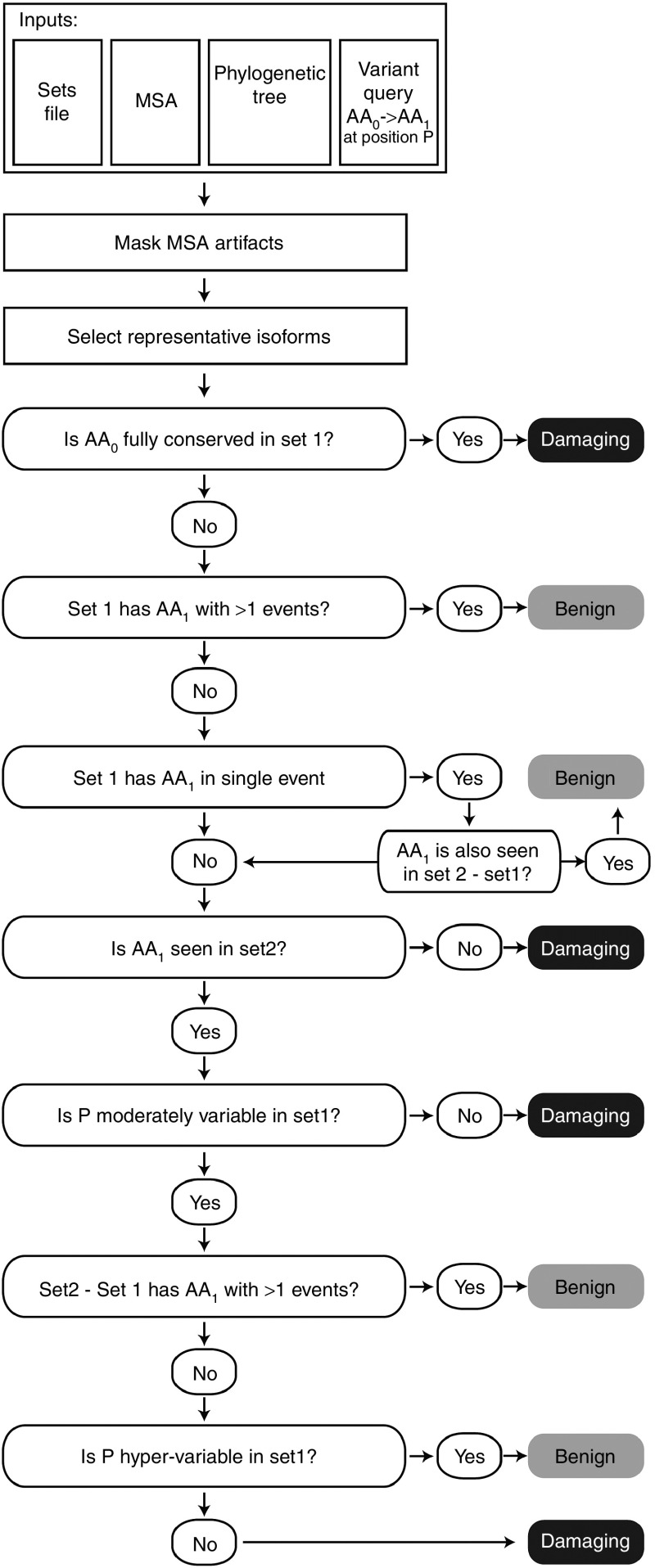
Methods: We identified major events in NPC1 evolution and revealed and compared orthologs and paralogs of the human NPC1 gene through phylogenetic and protein sequence analyses. We predicted whether an amino acid substitution affects protein function by reducing the organism's fitness.
Results: Removing the paralogs and distant homologs improved the overall performance of categorizing disease-causing and benign amino acid substitutions.
Conclusion: The results show that a thorough evolutionary analysis followed by identification of orthologs improves the accuracy in predicting disease-causing missense mutations. We anticipate that this approach will be used as a reference in the interpretation of variants in other genetic diseases as well.Genet Med 18 10, 1029-1036.
Figures

References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
