

Using single nuclei for RNA-seq to capture the transcriptome of postmortem neurons
- PMID: 26890679
- PMCID: PMC4941947
- DOI: 10.1038/nprot.2016.015
Using single nuclei for RNA-seq to capture the transcriptome of postmortem neurons
Abstract
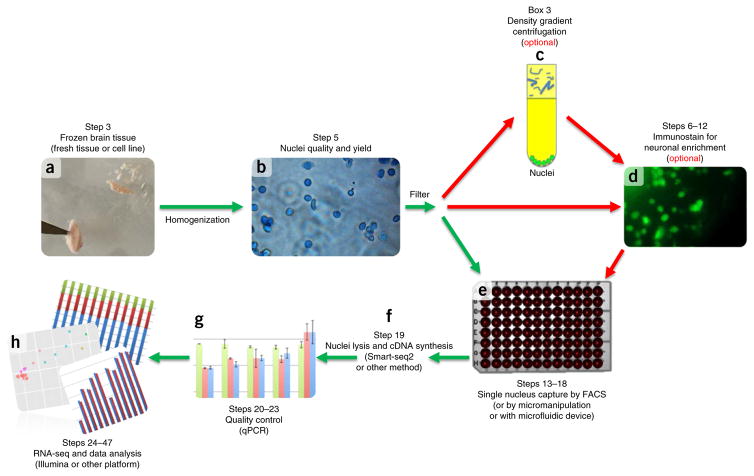
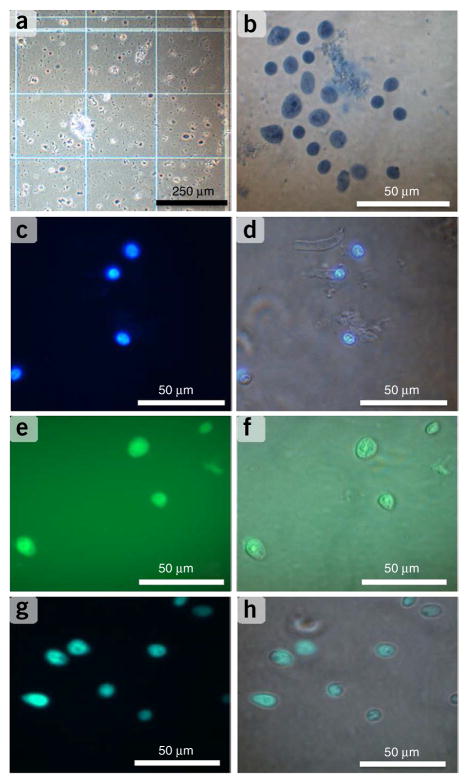
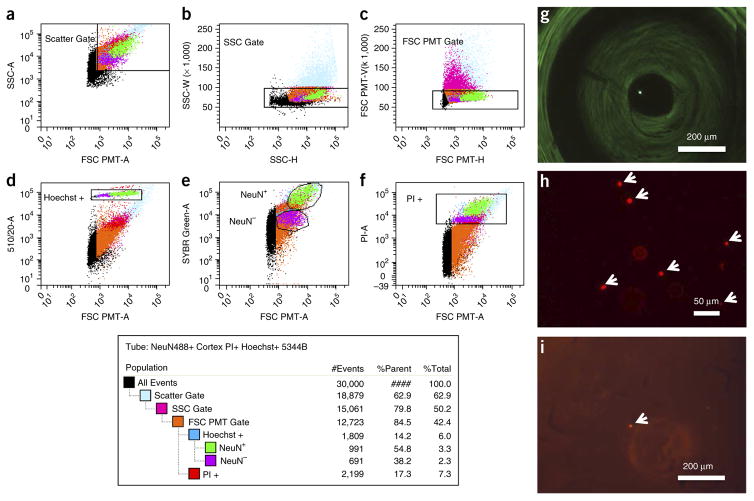
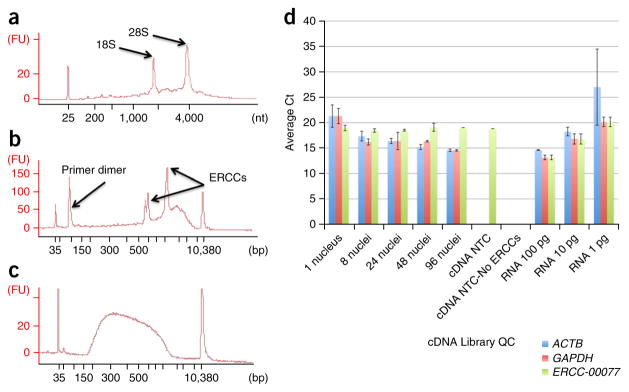
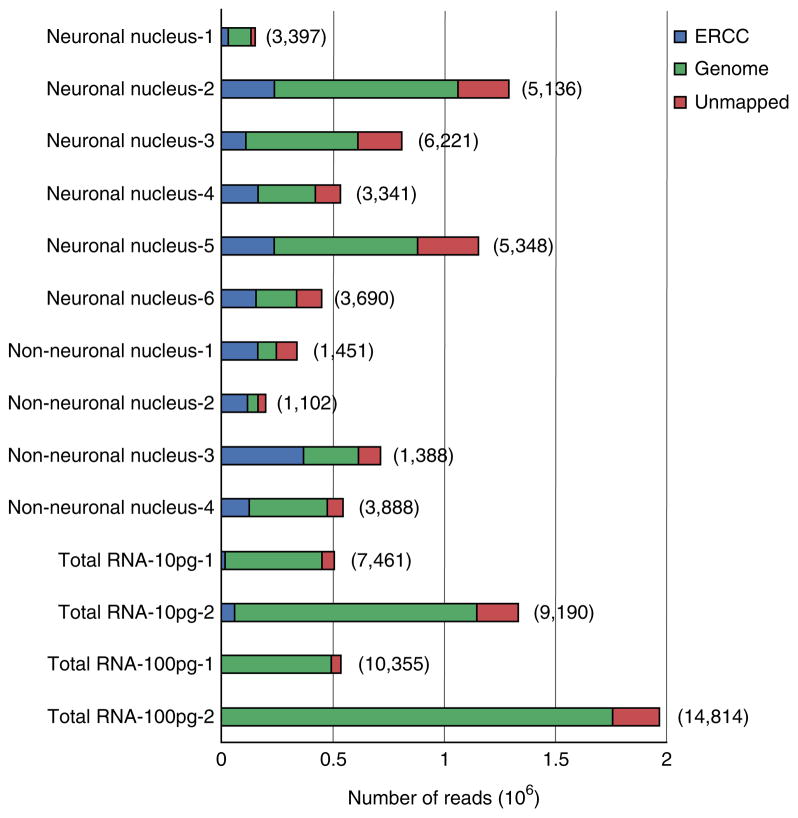
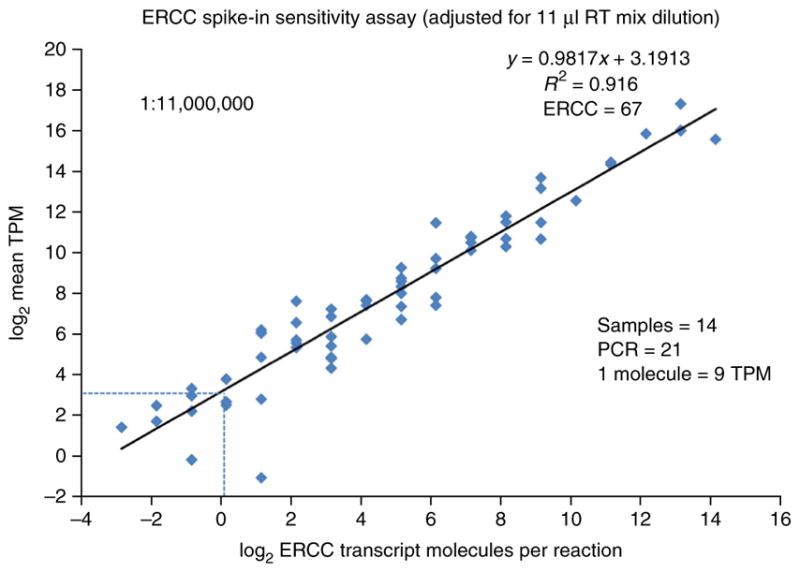
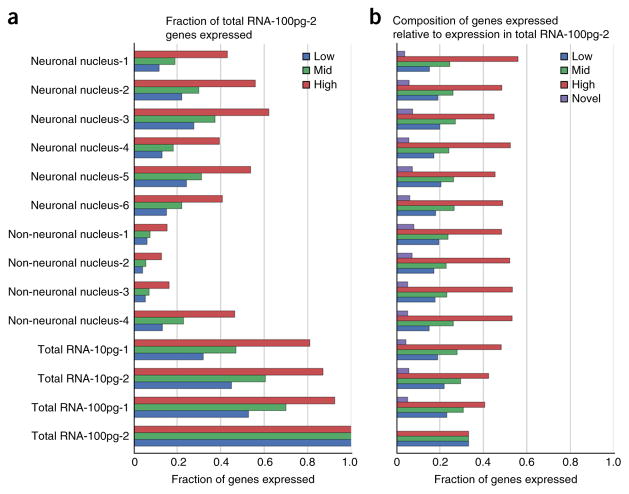
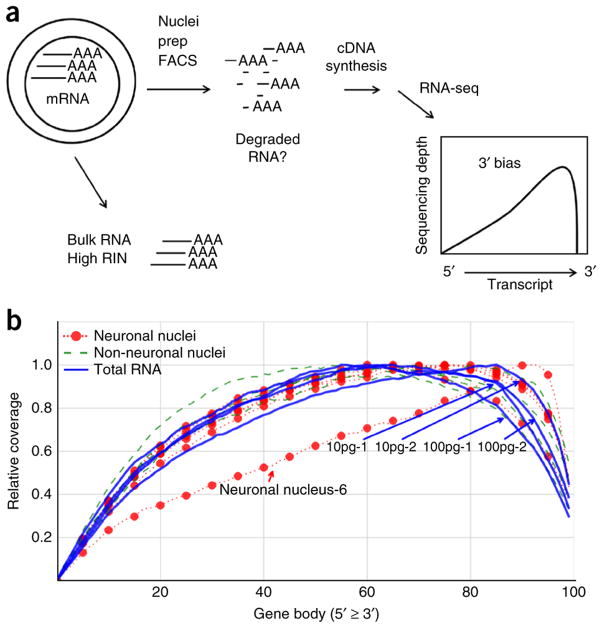
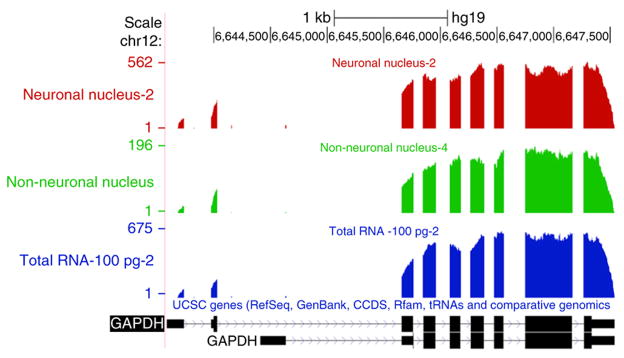
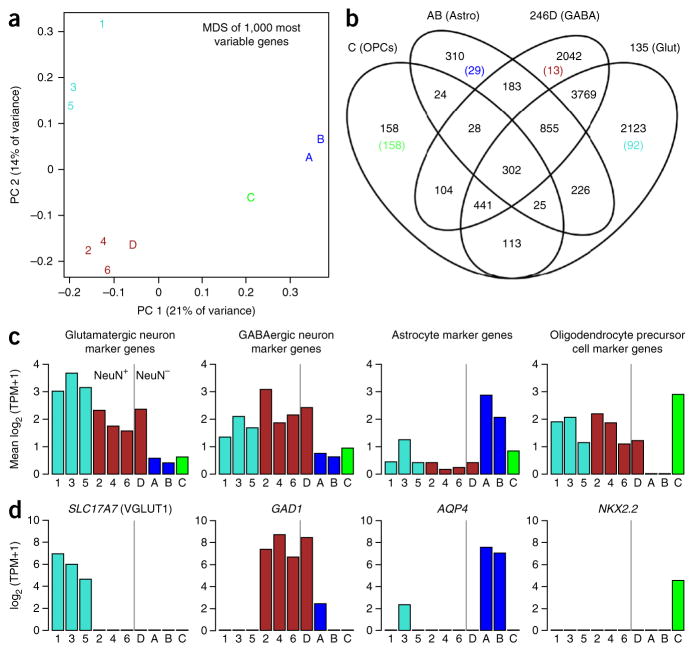
A protocol is described for sequencing the transcriptome of a cell nucleus. Nuclei are isolated from specimens and sorted by FACS, cDNA libraries are constructed and RNA-seq is performed, followed by data analysis. Some steps follow published methods (Smart-seq2 for cDNA synthesis and Nextera XT barcoded library preparation) and are not described in detail here. Previous single-cell approaches for RNA-seq from tissues include cell dissociation using protease treatment at 30 °C, which is known to alter the transcriptome. We isolate nuclei at 4 °C from tissue homogenates, which cause minimal damage. Nuclear transcriptomes can be obtained from postmortem human brain tissue stored at -80 °C, making brain archives accessible for RNA-seq from individual neurons. The method also allows investigation of biological features unique to nuclei, such as enrichment of certain transcripts and precursors of some noncoding RNAs. By following this procedure, it takes about 4 d to construct cDNA libraries that are ready for sequencing.
Conflict of interest statement
Figures
References
-
- Hashimshony T, Wagner F, Sher N, Yanai I. CEL-Seq: single-cell RNA-seq by multiplexed linear amplification. Cell Rep. 2012;2:666–673. - PubMed
-
- Kurimoto K, Yabuta Y, Ohinata Y, Saitou M. Global single-cell cDNA amplification to provide a template for representative high-density oligonucleotide microarray analysis. Nat Protoc. 2007;2:739–752. - PubMed
-
- Picelli S, et al. Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc. 2014;9:171–181. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
