

SNP discovery and genetic mapping using genotyping by sequencing of whole genome genomic DNA from a pea RIL population
- PMID: 26892170
- PMCID: PMC4758021
- DOI: 10.1186/s12864-016-2447-2
SNP discovery and genetic mapping using genotyping by sequencing of whole genome genomic DNA from a pea RIL population
Abstract
Background: Progress in genetics and breeding in pea still suffers from the limited availability of molecular resources. SNP markers that can be identified through affordable sequencing processes, without the need for prior genome reduction or a reference genome to assemble sequencing data would allow the discovery and genetic mapping of thousands of molecular markers. Such an approach could significantly speed up genetic studies and marker assisted breeding for non-model species.
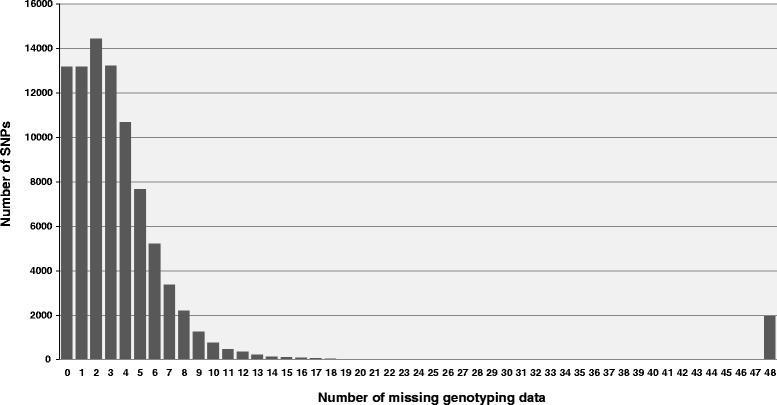
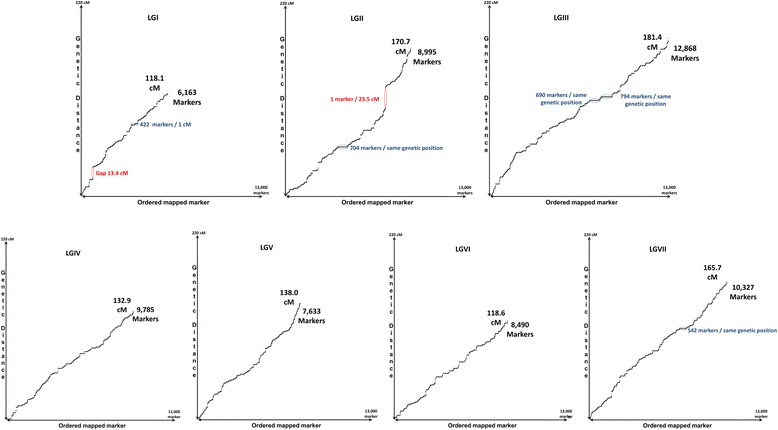
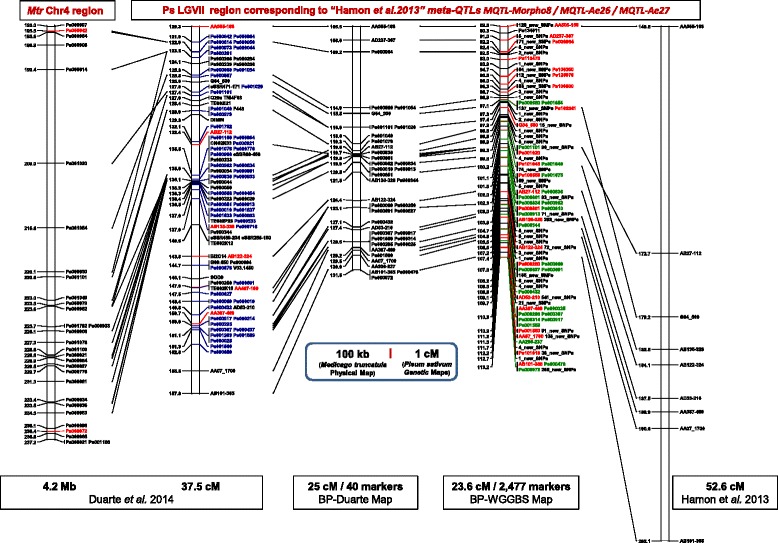
Results: A total of 419,024 SNPs were discovered using HiSeq whole genome sequencing of four pea lines, followed by direct identification of SNP markers without assembly using the discoSnp tool. Subsequent filtering led to the identification of 131,850 highly designable SNPs, polymorphic between at least two of the four pea lines. A subset of 64,754 SNPs was called and genotyped by short read sequencing on a subpopulation of 48 RILs from the cross 'Baccara' x 'PI180693'. This data was used to construct a WGGBS-derived pea genetic map comprising 64,263 markers. This map is collinear with previous pea consensus maps and therefore with the Medicago truncatula genome. Sequencing of four additional pea lines showed that 33 % to 64 % of the mapped SNPs, depending on the pairs of lines considered, are polymorphic and can therefore be useful in other crosses. The subsequent genotyping of a subset of 1000 SNPs, chosen for their mapping positions using a KASP™ assay, showed that almost all generated SNPs are highly designable and that most (95 %) deliver highly qualitative genotyping results. Using rather low sequencing coverages in SNP discovery and in SNP inferring did not hinder the identification of hundreds of thousands of high quality SNPs.
Conclusions: The development and optimization of appropriate tools in SNP discovery and genetic mapping have allowed us to make available a massive new genomic resource in pea. It will be useful for both fine mapping within chosen QTL confidence intervals and marker assisted breeding for important traits in pea improvement.
Figures
References
-
- Poland JA, Rife TW. Genotyping-by-sequencing for plant breeding and genetics. Plant Genome. 2012;5(3):92–102. doi: 10.3835/plantgenome2012.05.0005. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
