

Macrophage Phenotype and Function in Different Stages of Atherosclerosis
- PMID: 26892964
- PMCID: PMC4762068
- DOI: 10.1161/CIRCRESAHA.115.306256
Macrophage Phenotype and Function in Different Stages of Atherosclerosis
Abstract
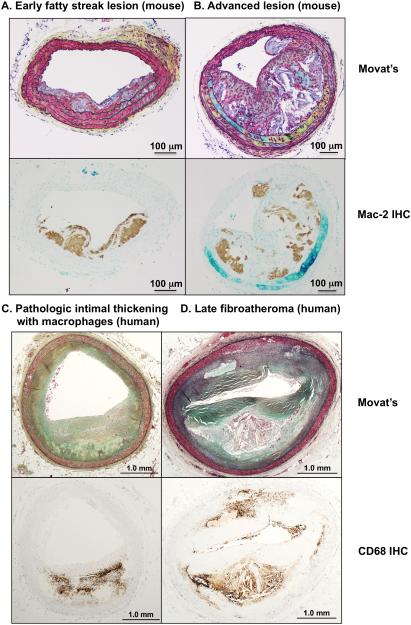
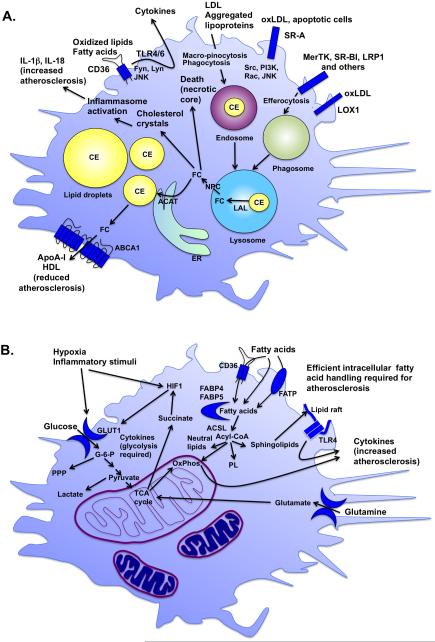
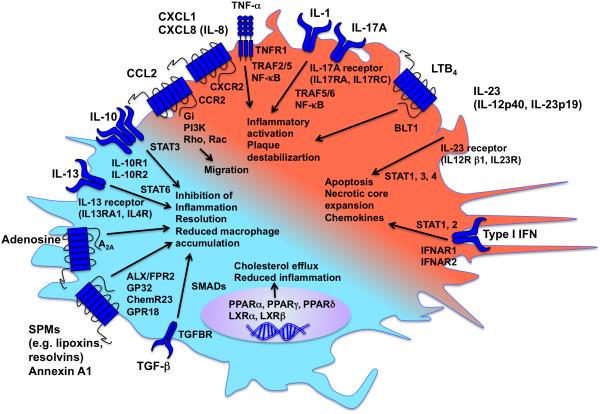
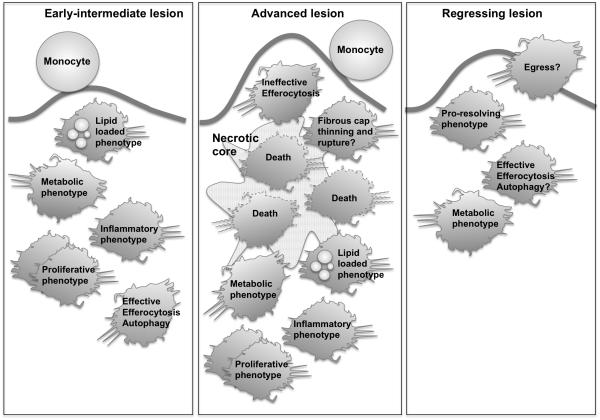
The remarkable plasticity and plethora of biological functions performed by macrophages have enticed scientists to study these cells in relation to atherosclerosis for >50 years, and major discoveries continue to be made today. It is now understood that macrophages play important roles in all stages of atherosclerosis, from initiation of lesions and lesion expansion, to necrosis leading to rupture and the clinical manifestations of atherosclerosis, to resolution and regression of atherosclerotic lesions. Lesional macrophages are derived primarily from blood monocytes, although recent research has shown that lesional macrophage-like cells can also be derived from smooth muscle cells. Lesional macrophages take on different phenotypes depending on their environment and which intracellular signaling pathways are activated. Rather than a few distinct populations of macrophages, the phenotype of the lesional macrophage is more complex and likely changes during the different phases of atherosclerosis and with the extent of lipid and cholesterol loading, activation by a plethora of receptors, and metabolic state of the cells. These different phenotypes allow the macrophage to engulf lipids, dead cells, and other substances perceived as danger signals; efflux cholesterol to high-density lipoprotein; proliferate and migrate; undergo apoptosis and death; and secrete a large number of inflammatory and proresolving molecules. This review article, part of the Compendium on Atherosclerosis, discusses recent advances in our understanding of lesional macrophage phenotype and function in different stages of atherosclerosis. With the increasing understanding of the roles of lesional macrophages, new research areas and treatment strategies are beginning to emerge.
Keywords: atherosclerosis; cholesterol; foam cells; macrophage; phenotype.
© 2016 American Heart Association, Inc.
Figures
References
-
- Ross R. Atherosclerosis--an inflammatory disease. N Engl J Med. 1999;340:115–126. - PubMed
-
- Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105:1135–1143. - PubMed
-
- Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–1695. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- HHSN268201000045C/HL/NHLBI NIH HHS/United States
- P01HL092969/HL/NHLBI NIH HHS/United States
- R01HL126028/HL/NHLBI NIH HHS/United States
- R01 HL107497/HL/NHLBI NIH HHS/United States
- HHSN268201000045C/PHS HHS/United States
- R01 HL062887/HL/NHLBI NIH HHS/United States
- R01 HL126028/HL/NHLBI NIH HHS/United States
- DP3 DK108209/DK/NIDDK NIH HHS/United States
- R01 HL075662/HL/NHLBI NIH HHS/United States
- R01HL107497/HL/NHLBI NIH HHS/United States
- P30DK017047/DK/NIDDK NIH HHS/United States
- R01HL062887/HL/NHLBI NIH HHS/United States
- P01 HL092969/HL/NHLBI NIH HHS/United States
- DP3DK108209/DK/NIDDK NIH HHS/United States
- P30 DK017047/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
