

Brain Insulin Signaling Is Increased in Insulin-Resistant States and Decreases in FOXOs and PGC-1α and Increases in Aβ1-40/42 and Phospho-Tau May Abet Alzheimer Development
- PMID: 26895791
- PMCID: PMC4915579
- DOI: 10.2337/db15-1428
Brain Insulin Signaling Is Increased in Insulin-Resistant States and Decreases in FOXOs and PGC-1α and Increases in Aβ1-40/42 and Phospho-Tau May Abet Alzheimer Development
Abstract
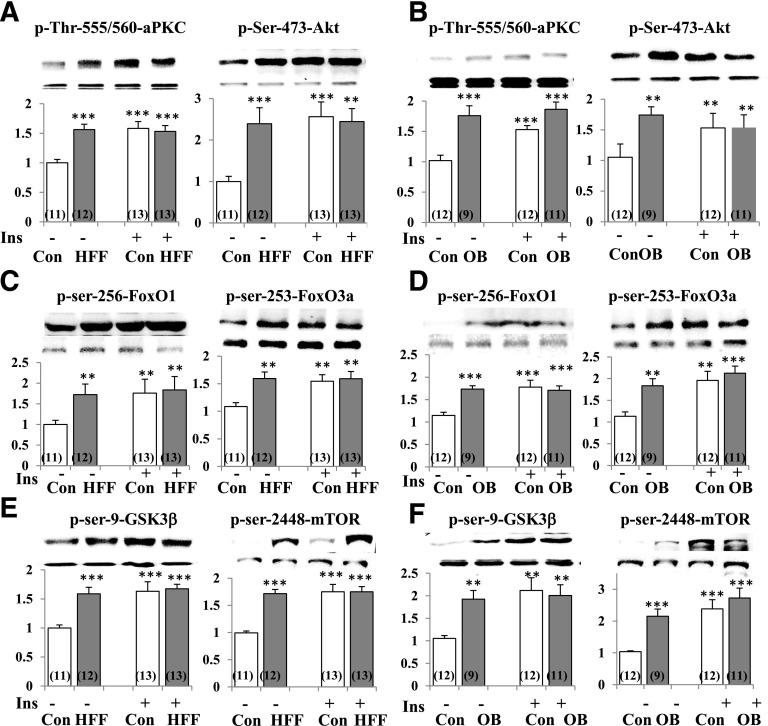
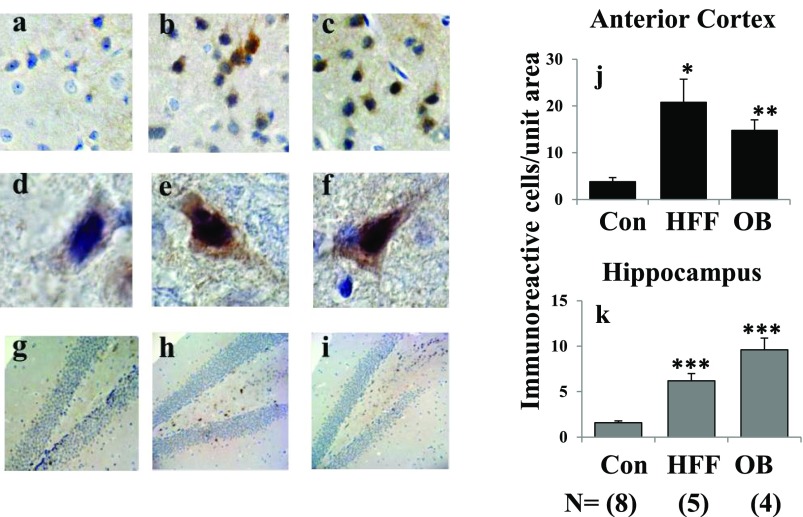
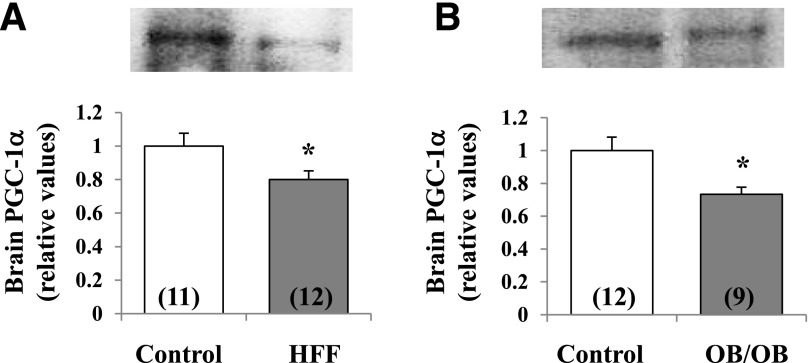
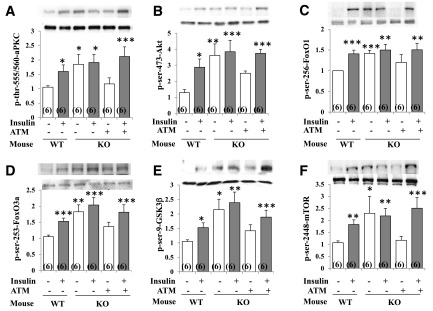
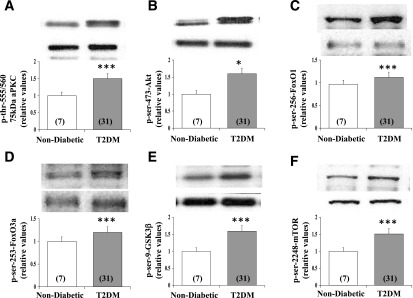
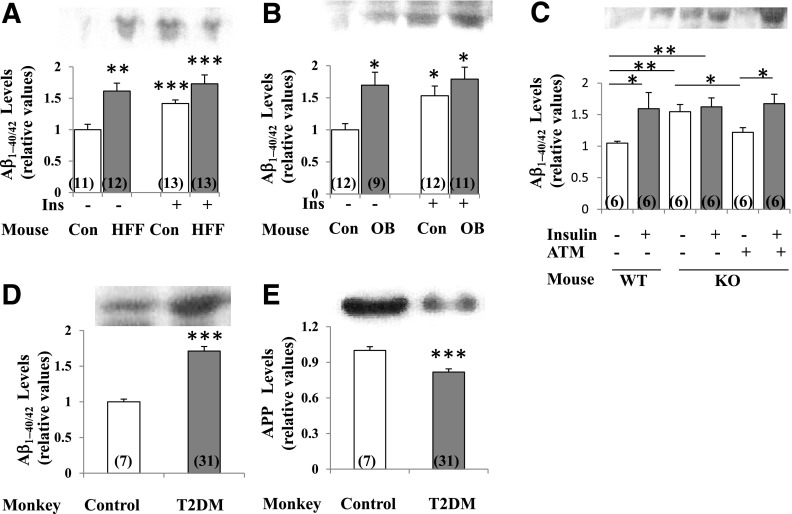
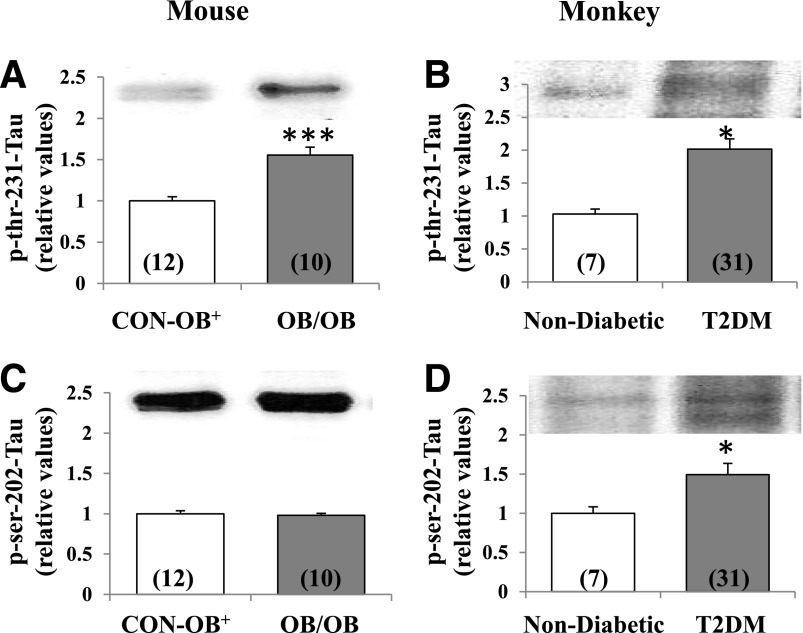
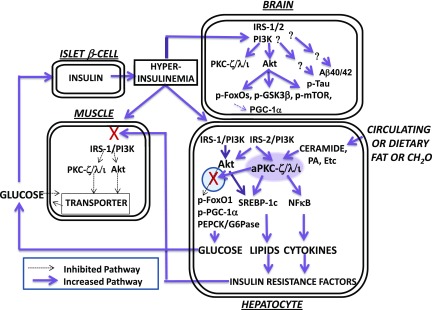
Increased coexistence of Alzheimer disease (AD) and type 2 diabetes mellitus (T2DM) suggests that insulin resistance abets neurodegenerative processes, but linkage mechanisms are obscure. Here, we examined insulin signaling factors in brains of insulin-resistant high-fat-fed mice, ob/ob mice, mice with genetically impaired muscle glucose transport, and monkeys with diet-dependent long-standing obesity/T2DM. In each model, the resting/basal activities of insulin-regulated brain protein kinases, Akt and atypical protein kinase C (aPKC), were maximally increased. Moreover, Akt hyperactivation was accompanied by hyperphosphorylation of substrates glycogen synthase kinase-3β and mammalian target of rapamycin and FOXO proteins FOXO1, FOXO3A, and FOXO4 and decreased peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α) expression. Akt hyperactivation was confirmed in individual neurons of anterocortical and hippocampal regions that house cognition/memory centers. Remarkably, β-amyloid (Aβ1-40/42) peptide levels were as follows: increased in the short term by insulin in normal mice, increased basally in insulin-resistant mice and monkeys, and accompanied by diminished amyloid precursor protein in monkeys. Phosphorylated tau levels were increased in ob/ob mice and T2DM monkeys. Importantly, with correction of hyperinsulinemia by inhibition of hepatic aPKC and improvement in systemic insulin resistance, brain insulin signaling normalized. As FOXOs and PGC-1α are essential for memory and long-term neuronal function and regeneration and as Aβ1-40/42 and phospho-tau may increase interneuronal plaques and intraneuronal tangles, presently observed aberrations in hyperinsulinemic states may participate in linking insulin resistance to AD.
© 2016 by the American Diabetes Association. Readers may use this article as long as the work is properly cited, the use is educational and not for profit, and the work is not altered.
Figures
References
-
- Ford ES, Giles WH, Dietz WH. Prevalence of the metabolic syndrome among US adults: findings from the third National Health and Nutrition Examination Survey. JAMA 2002;287:356–359 - PubMed
-
- Cukierman T, Gerstein HC, Williamson JD. Cognitive decline and dementia in diabetes—systematic overview of prospective observational studies. Diabetologia 2005;48:2460–2469 - PubMed
-
- Ott A, Stolk RP, Hofman A, van Harskamp F, Grobbee DE, Breteler MMB. Association of diabetes mellitus and dementia: the Rotterdam Study. Diabetologia 1996;39:1392–1397 - PubMed
-
- Janson J, Laedtke T, Parisi JE, O’Brien P, Petersen RC, Butler PC. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes 2004;53:474–481 - PubMed
-
- Frölich L, Blum-Degen D, Bernstein HG, et al. . Brain insulin and insulin receptors in aging and sporadic Alzheimer’s disease. J Neural Transm (Vienna) 1998;105:423–438 - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
