

Reversal of epigenetic promoter silencing in Friedreich ataxia by a class I histone deacetylase inhibitor
- PMID: 26896803
- PMCID: PMC4914082
- DOI: 10.1093/nar/gkw107
Reversal of epigenetic promoter silencing in Friedreich ataxia by a class I histone deacetylase inhibitor
Abstract
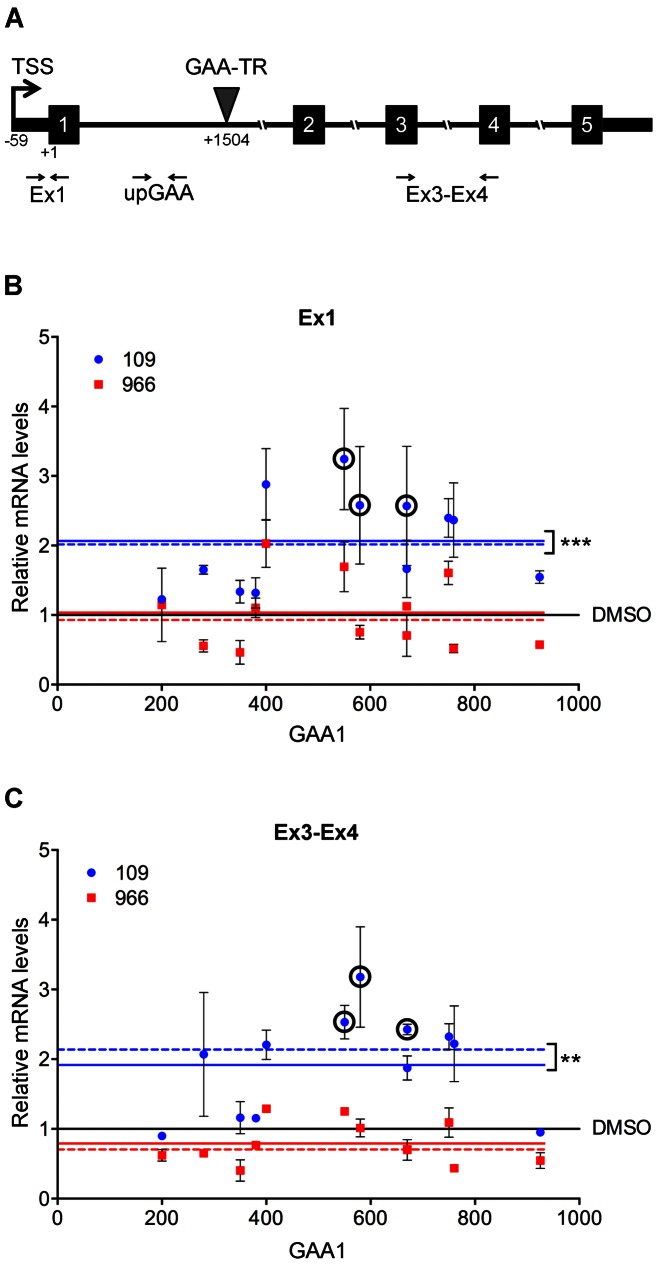
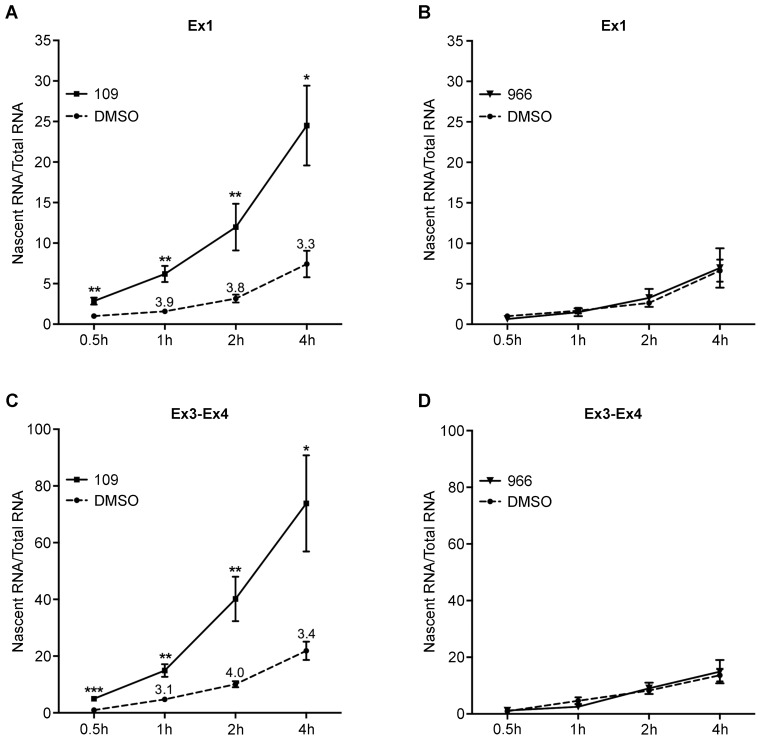
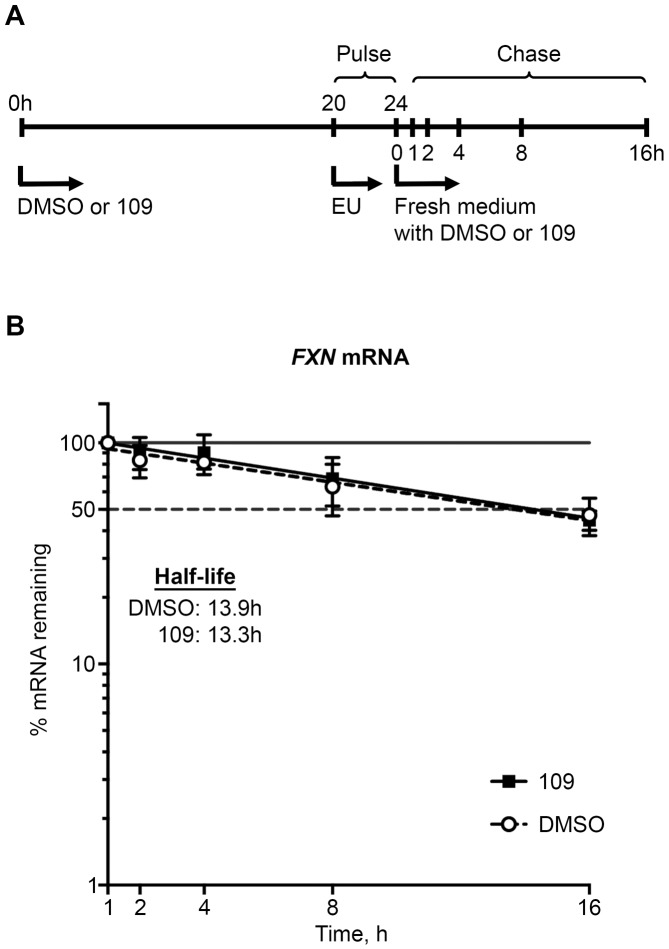
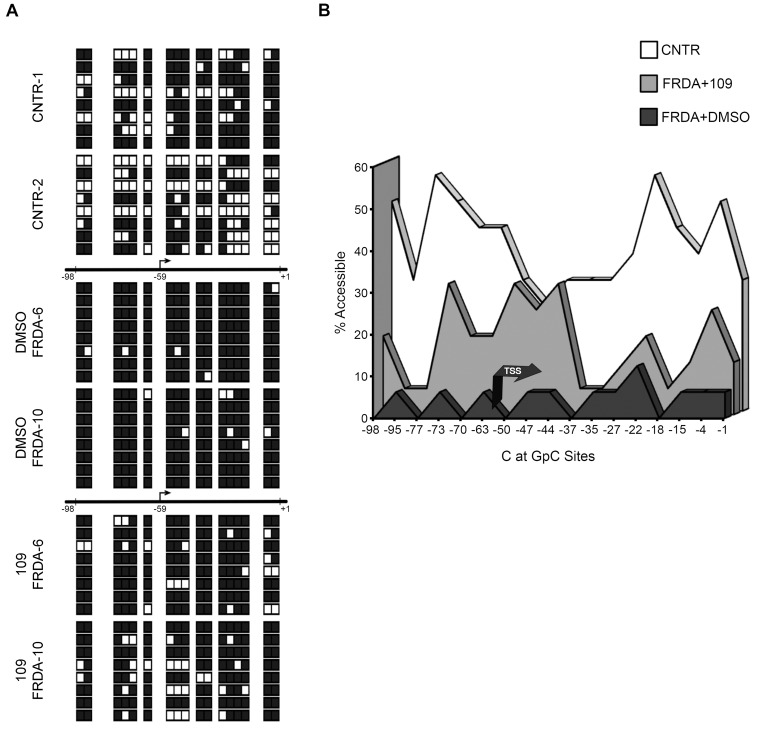
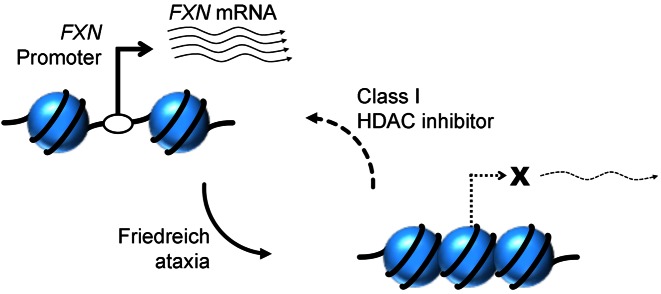
Friedreich ataxia, the most prevalent inherited ataxia, is caused by an expanded GAA triplet-repeat sequence in intron 1 of the FXN gene. Repressive chromatin spreads from the expanded GAA triplet-repeat sequence to cause epigenetic silencing of the FXN promoter via altered nucleosomal positioning and reduced chromatin accessibility. Indeed, deficient transcriptional initiation is the predominant cause of transcriptional deficiency in Friedreich ataxia. Treatment with 109, a class I histone deacetylase (HDAC) inhibitor, resulted in increased level of FXN transcript both upstream and downstream of the expanded GAA triplet-repeat sequence, without any change in transcript stability, suggesting that it acts via improvement of transcriptional initiation. Quantitative analysis of transcriptional initiation via metabolic labeling of nascent transcripts in patient-derived cells revealed a >3-fold increase (P < 0.05) in FXN promoter function. A concomitant 3-fold improvement (P < 0.001) in FXN promoter structure and chromatin accessibility was observed via Nucleosome Occupancy and Methylome Sequencing, a high-resolution in vivo footprint assay for detecting nucleosome occupancy in individual chromatin fibers. No such improvement in FXN promoter function or structure was observed upon treatment with a chemically-related inactive compound (966). Thus epigenetic promoter silencing in Friedreich ataxia is reversible, and the results implicate class I HDACs in repeat-mediated promoter silencing.
© The Author(s) 2016. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures

Similar articles
-
Altered nucleosome positioning at the transcription start site and deficient transcriptional initiation in Friedreich ataxia.J Biol Chem. 2014 May 30;289(22):15194-202. doi: 10.1074/jbc.M114.566414. Epub 2014 Apr 15. J Biol Chem. 2014. PMID: 24737321 Free PMC article.
-
FXN Promoter Silencing in the Humanized Mouse Model of Friedreich Ataxia.PLoS One. 2015 Sep 22;10(9):e0138437. doi: 10.1371/journal.pone.0138437. eCollection 2015. PLoS One. 2015. PMID: 26393353 Free PMC article.
-
Epigenetic promoter silencing in Friedreich ataxia is dependent on repeat length.Ann Neurol. 2014 Oct;76(4):522-8. doi: 10.1002/ana.24249. Epub 2014 Aug 30. Ann Neurol. 2014. PMID: 25112975 Free PMC article.
-
Molecular Mechanisms and Therapeutics for the GAA·TTC Expansion Disease Friedreich Ataxia.Neurotherapeutics. 2019 Oct;16(4):1032-1049. doi: 10.1007/s13311-019-00764-x. Neurotherapeutics. 2019. PMID: 31317428 Free PMC article. Review.
-
Analysis of Putative Epigenetic Regulatory Elements in the FXN Genomic Locus.Int J Mol Sci. 2020 May 12;21(10):3410. doi: 10.3390/ijms21103410. Int J Mol Sci. 2020. PMID: 32408537 Free PMC article. Review.
Cited by
-
Selected Histone Deacetylase Inhibitors Reverse the Frataxin Transcriptional Defect in a Novel Friedreich's Ataxia Induced Pluripotent Stem Cell-Derived Neuronal Reporter System.Front Neurosci. 2022 Feb 23;16:836476. doi: 10.3389/fnins.2022.836476. eCollection 2022. Front Neurosci. 2022. PMID: 35281493 Free PMC article.
-
Friedreich Ataxia: current status and future prospects.Cerebellum Ataxias. 2017 Apr 7;4:4. doi: 10.1186/s40673-017-0062-x. eCollection 2017. Cerebellum Ataxias. 2017. PMID: 28405347 Free PMC article. Review.
-
Depending on the stress, histone deacetylase inhibitors act as heat shock protein co-inducers in motor neurons and potentiate arimoclomol, exerting neuroprotection through multiple mechanisms in ALS models.Cell Stress Chaperones. 2020 Jan;25(1):173-191. doi: 10.1007/s12192-019-01064-1. Epub 2020 Jan 3. Cell Stress Chaperones. 2020. PMID: 31900865 Free PMC article.
-
HMTase Inhibitors as a Potential Epigenetic-Based Therapeutic Approach for Friedreich's Ataxia.Front Genet. 2020 Jun 5;11:584. doi: 10.3389/fgene.2020.00584. eCollection 2020. Front Genet. 2020. PMID: 32582297 Free PMC article.
-
Epigenetic Heterogeneity in Friedreich Ataxia Underlies Variable FXN Reactivation.Front Neurosci. 2021 Nov 25;15:752921. doi: 10.3389/fnins.2021.752921. eCollection 2021. Front Neurosci. 2021. PMID: 34899161 Free PMC article.
References
-
- Saveliev A., Everett C., Sharpe T., Webster Z., Festenstein R. DNA triplet repeats mediate heterochromatin-protein-1-sensitive variegated gene silencing. Nature. 2003;422:909–913. - PubMed
-
- Campuzano V., Montermini L., Molto M.D., Pianese L., Cossee M., Cavalcanti F., Monros E., Rodius F., Duclos F., Monticelli A., et al. Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271:1423–1427. - PubMed
-
- Herman D., Jenssen K., Burnett R., Soragni E., Perlman S.L., Gottesfeld J.M. Histone deacetylase inhibitors reverse gene silencing in Friedreich's ataxia. Nat. Chem. Biol. 2006;2:551–558. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
