

Ensembl comparative genomics resources
- PMID: 26896847
- PMCID: PMC4761110
- DOI: 10.1093/database/bav096
Ensembl comparative genomics resources
Erratum in
-
Ensembl comparative genomics resources.Database (Oxford). 2016 May 2;2016:baw053. doi: 10.1093/database/baw053. Print 2016. Database (Oxford). 2016. PMID: 27141089 Free PMC article. No abstract available.
Abstract
Evolution provides the unifying framework with which to understand biology. The coherent investigation of genic and genomic data often requires comparative genomics analyses based on whole-genome alignments, sets of homologous genes and other relevant datasets in order to evaluate and answer evolutionary-related questions. However, the complexity and computational requirements of producing such data are substantial: this has led to only a small number of reference resources that are used for most comparative analyses. The Ensembl comparative genomics resources are one such reference set that facilitates comprehensive and reproducible analysis of chordate genome data. Ensembl computes pairwise and multiple whole-genome alignments from which large-scale synteny, per-base conservation scores and constrained elements are obtained. Gene alignments are used to define Ensembl Protein Families, GeneTrees and homologies for both protein-coding and non-coding RNA genes. These resources are updated frequently and have a consistent informatics infrastructure and data presentation across all supported species. Specialized web-based visualizations are also available including synteny displays, collapsible gene tree plots, a gene family locator and different alignment views. The Ensembl comparative genomics infrastructure is extensively reused for the analysis of non-vertebrate species by other projects including Ensembl Genomes and Gramene and much of the information here is relevant to these projects. The consistency of the annotation across species and the focus on vertebrates makes Ensembl an ideal system to perform and support vertebrate comparative genomic analyses. We use robust software and pipelines to produce reference comparative data and make it freely available. Database URL: http://www.ensembl.org.
© The Author(s) 2016. Published by Oxford University Press.
Figures
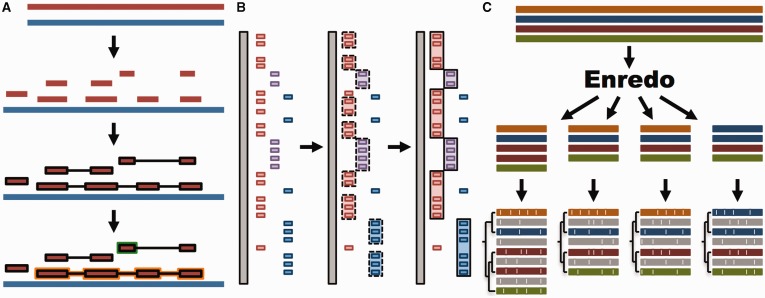
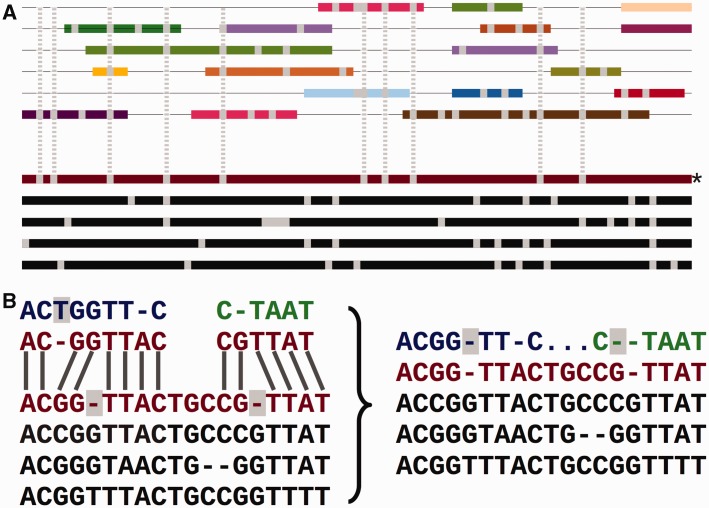
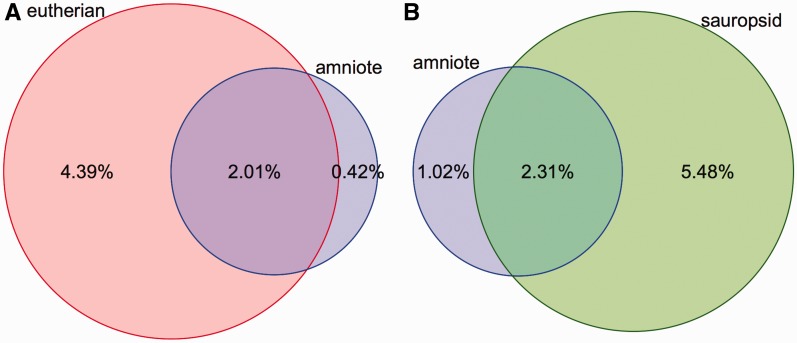
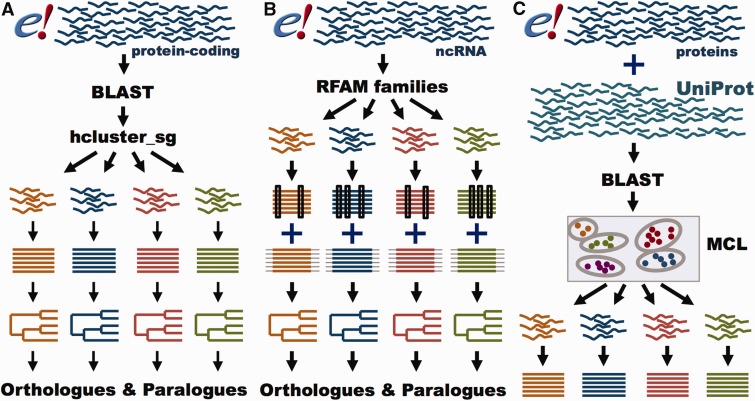
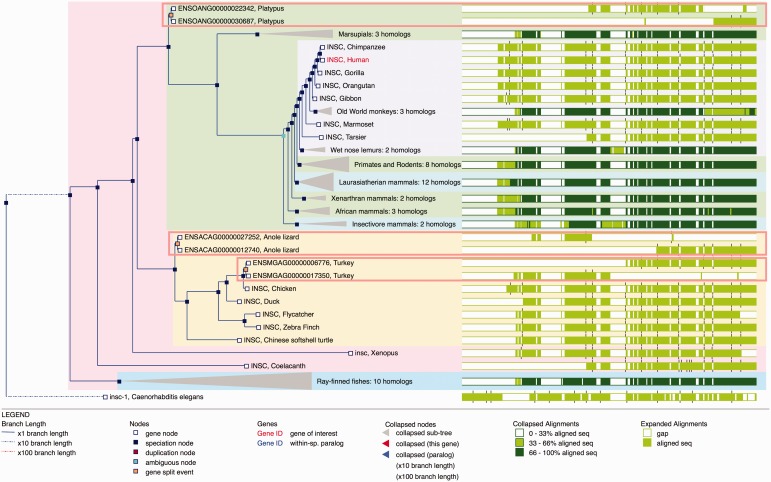
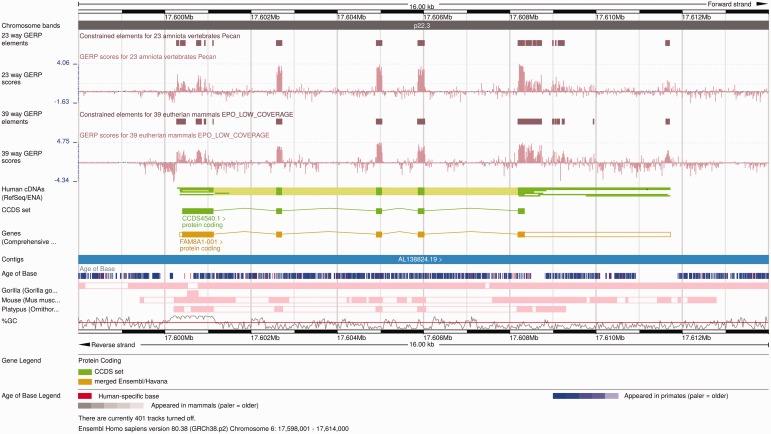
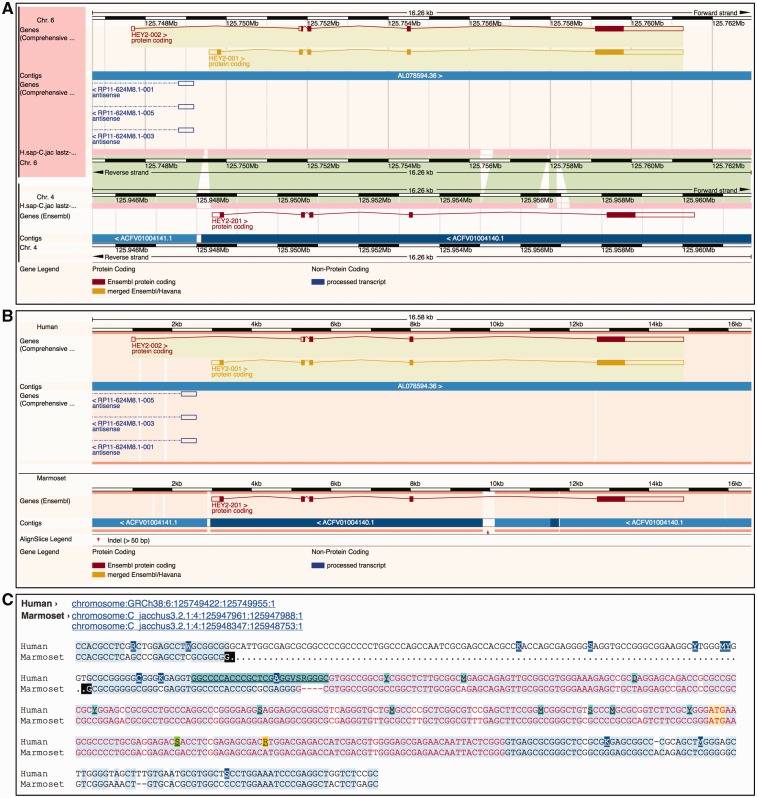
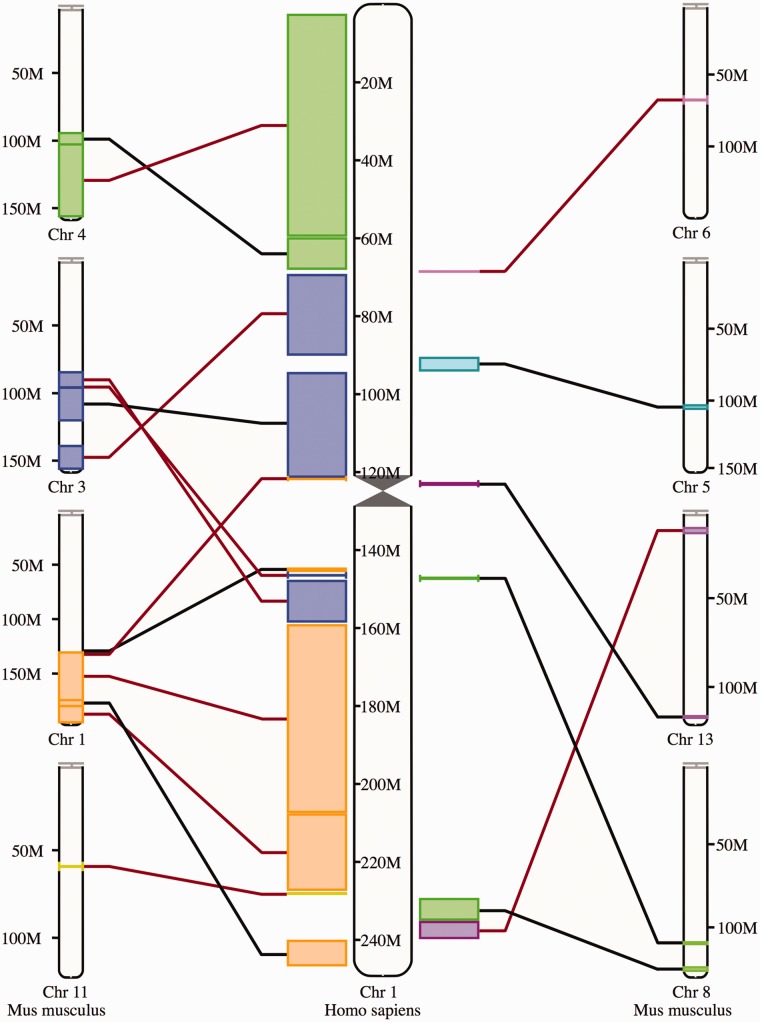
References
Publication types
MeSH terms
Substances
Grants and funding
- BB/I025360/2/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- U01HG004695/HG/NHGRI NIH HHS/United States
- BB/I025360/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- WT095908/WT_/Wellcome Trust/United Kingdom
- U41 HG006104/HG/NHGRI NIH HHS/United States
- BB/I025506/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- U01 HG004695/HG/NHGRI NIH HHS/United States
- WT098051/WT_/Wellcome Trust/United Kingdom
- U54 HG004563/HG/NHGRI NIH HHS/United States
- BB/E011640/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- U41HG006104/HG/NHGRI NIH HHS/United States
- U54HG004563/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
