

CD36 is a co-receptor for hepatitis C virus E1 protein attachment
- PMID: 26898231
- PMCID: PMC4761891
- DOI: 10.1038/srep21808
CD36 is a co-receptor for hepatitis C virus E1 protein attachment
Abstract
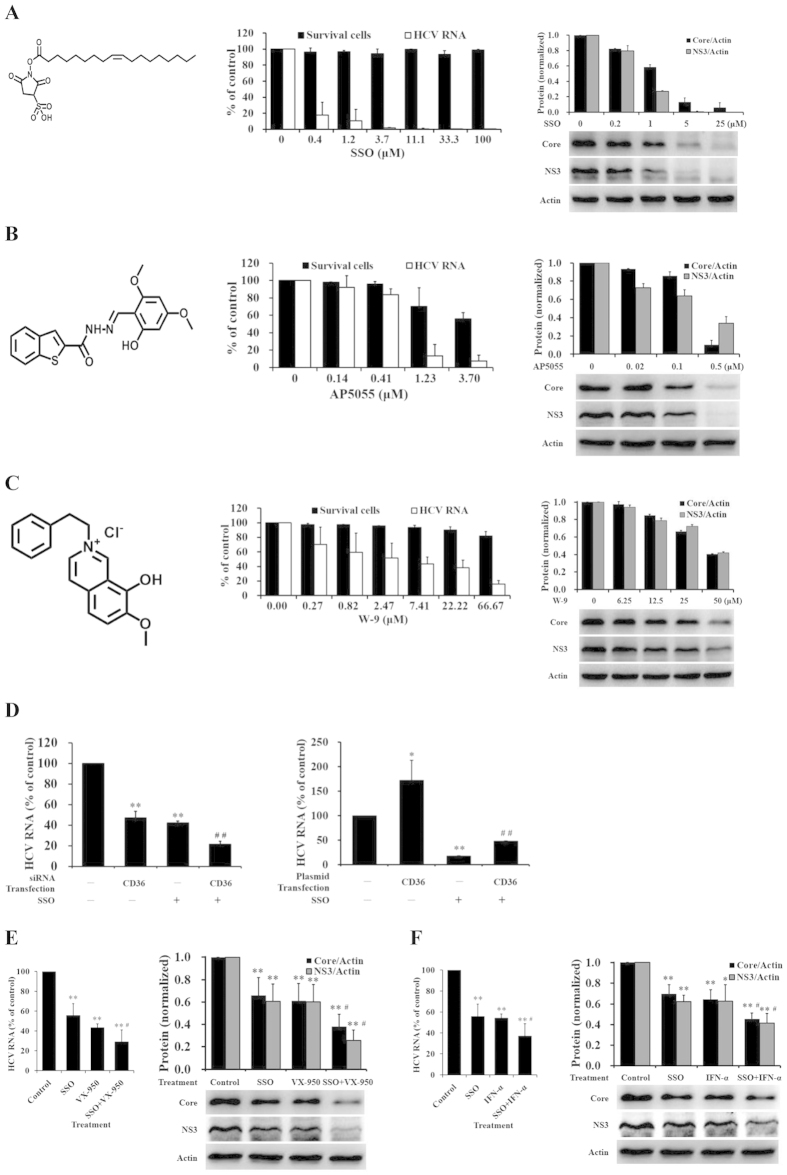
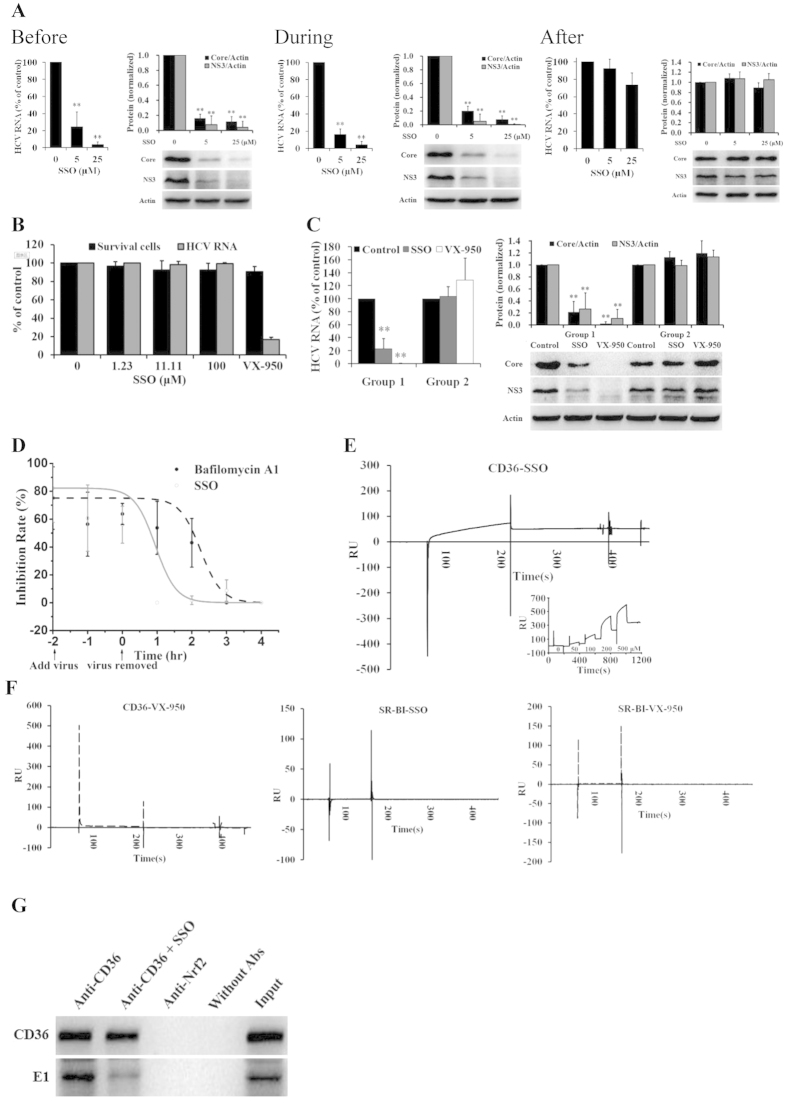
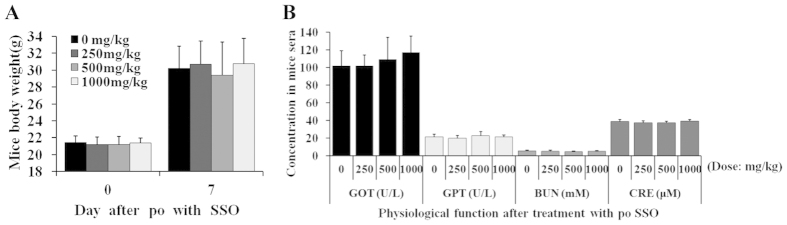
The cluster of differentiation 36 (CD36) is a membrane protein related to lipid metabolism. We show that HCV infection in vitro increased CD36 expression in either surface or soluble form. HCV attachment was facilitated through a direct interaction between CD36 and HCV E1 protein, causing enhanced entry and replication. The HCV co-receptor effect of CD36 was independent of that of SR-BI. CD36 monoclonal antibodies neutralized the effect of CD36 and reduced HCV replication. CD36 inhibitor sulfo-N-succinimidyl oleate (SSO), which directly bound CD36 but not SR-BI, significantly interrupted HCV entry, and therefore inhibited HCV replication. SSO's antiviral effect was seen only in HCV but not in other viruses. SSO in combination with known anti-HCV drugs showed additional inhibition against HCV. SSO was considerably safe in mice. Conclusively, CD36 interacts with HCV E1 and might be a co-receptor specific for HCV entry; thus, CD36 could be a potential drug target against HCV.
Figures



Similar articles
-
A Novel Inhibitor IDPP Interferes with Entry and Egress of HCV by Targeting Glycoprotein E1 in a Genotype-Specific Manner.Sci Rep. 2017 Mar 23;7:44676. doi: 10.1038/srep44676. Sci Rep. 2017. PMID: 28333153 Free PMC article.
-
Interferon alpha decreases expression of human scavenger receptor class BI, a possible HCV receptor in hepatocytes.Gut. 2008 May;57(5):664-71. doi: 10.1136/gut.2006.111443. Epub 2007 Nov 12. Gut. 2008. PMID: 17998316
-
(-)-Epigallocatechin-3-gallate is a new inhibitor of hepatitis C virus entry.Hepatology. 2012 Mar;55(3):720-9. doi: 10.1002/hep.24803. Hepatology. 2012. PMID: 22105803
-
[Research on hepatitis C virus entry inhibitor].Bing Du Xue Bao. 2015 Jan;31(1):97-105. Bing Du Xue Bao. 2015. PMID: 25997338 Review. Chinese.
-
Hepatitis C virus entry into hepatocytes: molecular mechanisms and targets for antiviral therapies.J Hepatol. 2011 Mar;54(3):566-76. doi: 10.1016/j.jhep.2010.10.014. Epub 2010 Nov 11. J Hepatol. 2011. PMID: 21146244 Review.
Cited by
-
From Structural Studies to HCV Vaccine Design.Viruses. 2021 May 4;13(5):833. doi: 10.3390/v13050833. Viruses. 2021. PMID: 34064532 Free PMC article. Review.
-
Determinants in the Ig Variable Domain of Human HAVCR1 (TIM-1) Are Required To Enhance Hepatitis C Virus Entry.J Virol. 2018 Feb 26;92(6):e01742-17. doi: 10.1128/JVI.01742-17. Print 2018 Mar 15. J Virol. 2018. PMID: 29321304 Free PMC article.
-
Infections at the nexus of metabolic-associated fatty liver disease.Arch Toxicol. 2021 Jul;95(7):2235-2253. doi: 10.1007/s00204-021-03069-1. Epub 2021 May 24. Arch Toxicol. 2021. PMID: 34027561 Free PMC article. Review.
-
Genome-Wide Transcriptome Analysis of CD36 Overexpression in HepG2.2.15 Cells to Explore Its Regulatory Role in Metabolism and the Hepatitis B Virus Life Cycle.PLoS One. 2016 Oct 17;11(10):e0164787. doi: 10.1371/journal.pone.0164787. eCollection 2016. PLoS One. 2016. PMID: 27749922 Free PMC article.
-
A Simple but Accurate Method for Evaluating Drug-Resistance in Infectious HCVcc System.Biomed Res Int. 2017;2017:1236801. doi: 10.1155/2017/1236801. Epub 2017 Aug 22. Biomed Res Int. 2017. PMID: 28904942 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
