

ER stress and the decline and fall of pancreatic beta cells in type 1 diabetes
- PMID: 26899404
- PMCID: PMC4900073
- DOI: 10.3109/03009734.2015.1135217
ER stress and the decline and fall of pancreatic beta cells in type 1 diabetes
Abstract
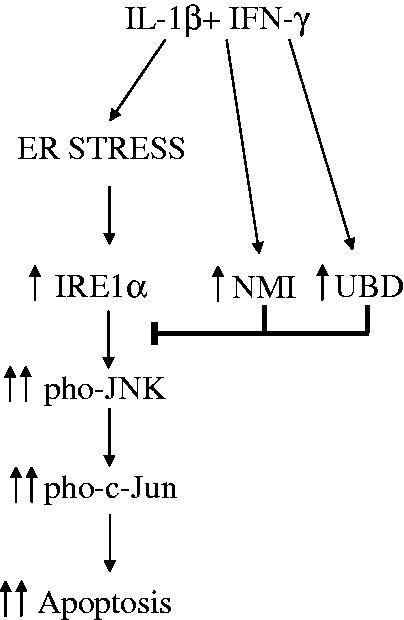
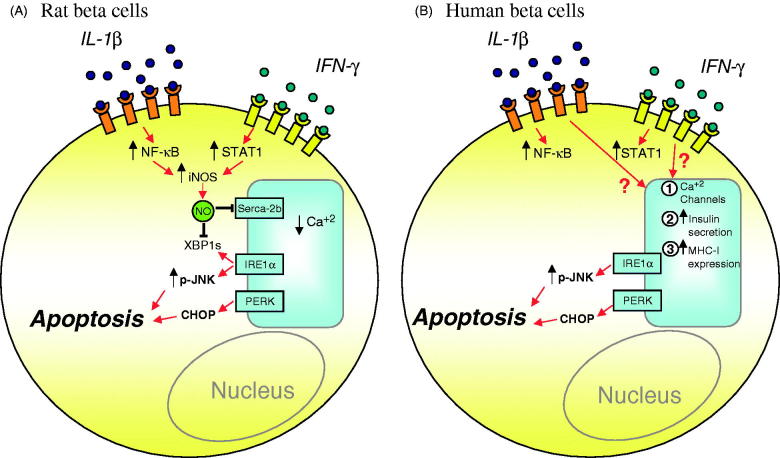
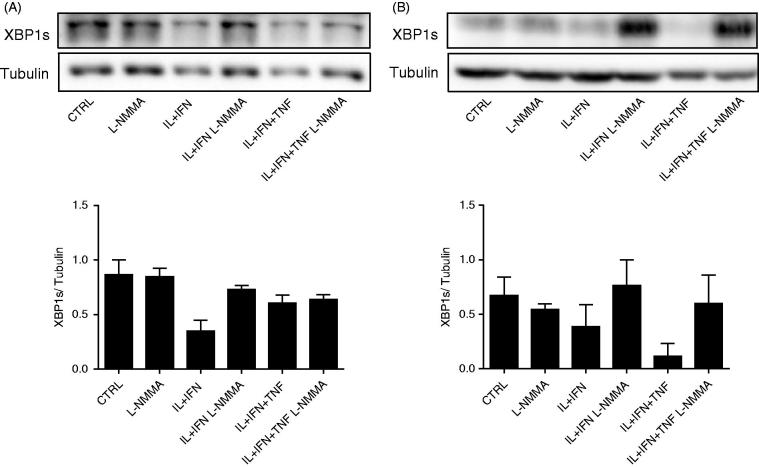
Components of the unfolded protein response (UPR) modulate beta cell inflammation and death in early type 1 diabetes (T1D). The UPR is a mechanism by which cells react to the accumulation of misfolded proteins in the endoplasmic reticulum (ER). It aims to restore cellular homeostasis, but in case of chronic or overwhelming ER stress the persistent activation of the UPR triggers apoptosis, contributing to the loss of beta cells in both T1D and type 2 diabetes. It remains to be determined how and why the transition from 'physiological' to 'pathological' UPR takes place. A key component of the UPR is the ER transmembrane protein IRE1α (inositol-requiring enzyme 1α). IRE1α activity is modulated by both intra-ER signals and by the formation of protein complexes at its cytosolic domain. The amplitude and duration of IRE1α signaling is critical for the transition between the adaptive and cell death programs, with particular relevance for the activation of the pro-apoptotic c-Jun N-terminal kinase (JNK) in beta cells. In the present review we discuss the available information on IRE1α-regulating proteins in beta cells and their downstream targets, and the important differences observed between cytokine-induced UPR in human and rodent beta cells.
Keywords: Apoptosis; ER stress; IRE1α; c-Jun N-terminal kinase; cytokines; type 1 diabetes.
Figures
Similar articles
-
Ubiquitin D Regulates IRE1α/c-Jun N-terminal Kinase (JNK) Protein-dependent Apoptosis in Pancreatic Beta Cells.J Biol Chem. 2016 Jun 3;291(23):12040-56. doi: 10.1074/jbc.M115.704619. Epub 2016 Apr 4. J Biol Chem. 2016. PMID: 27044747 Free PMC article.
-
Nicotinic acetylcholine receptor signaling regulates inositol-requiring enzyme 1α activation to protect β-cells against terminal unfolded protein response under irremediable endoplasmic reticulum stress.J Diabetes Investig. 2020 Jul;11(4):801-813. doi: 10.1111/jdi.13211. Epub 2020 Feb 17. J Diabetes Investig. 2020. PMID: 31925927 Free PMC article.
-
A combined "omics" approach identifies N-Myc interactor as a novel cytokine-induced regulator of IRE1 protein and c-Jun N-terminal kinase in pancreatic beta cells.J Biol Chem. 2014 Jul 25;289(30):20677-93. doi: 10.1074/jbc.M114.568808. J Biol Chem. 2014. PMID: 24936061 Free PMC article.
-
Molecular signal networks and regulating mechanisms of the unfolded protein response.J Zhejiang Univ Sci B. 2017 Jan.;18(1):1-14. doi: 10.1631/jzus.B1600043. J Zhejiang Univ Sci B. 2017. PMID: 28070992 Free PMC article. Review.
-
Endoplasmic reticulum stress, degeneration of pancreatic islet β-cells, and therapeutic modulation of the unfolded protein response in diabetes.Mol Metab. 2019 Sep;27S(Suppl):S60-S68. doi: 10.1016/j.molmet.2019.06.012. Mol Metab. 2019. PMID: 31500832 Free PMC article. Review.
Cited by
-
PEP-1-paraoxonase 1 fusion protein prevents cytokine-induced cell destruction and impaired insulin secretion in rat insulinoma cells.BMB Rep. 2018 Oct;51(10):538-543. doi: 10.5483/BMBRep.2018.51.10.181. BMB Rep. 2018. PMID: 30269741 Free PMC article.
-
PAHSAs attenuate immune responses and promote β cell survival in autoimmune diabetic mice.J Clin Invest. 2019 Aug 5;129(9):3717-3731. doi: 10.1172/JCI122445. eCollection 2019 Aug 5. J Clin Invest. 2019. PMID: 31380811 Free PMC article.
-
Pancreatic β-cells in type 1 and type 2 diabetes mellitus: different pathways to failure.Nat Rev Endocrinol. 2020 Jul;16(7):349-362. doi: 10.1038/s41574-020-0355-7. Epub 2020 May 12. Nat Rev Endocrinol. 2020. PMID: 32398822 Review.
-
Type I interferons and endoplasmic reticulum stress in health and disease.Int Rev Cell Mol Biol. 2020;350:63-118. doi: 10.1016/bs.ircmb.2019.10.004. Epub 2019 Nov 19. Int Rev Cell Mol Biol. 2020. PMID: 32138904 Free PMC article. Review.
-
Heat Shock Proteins as a Potential Therapeutic Target in the Treatment of Gestational Diabetes Mellitus: What We Know so Far.Int J Mol Sci. 2018 Oct 17;19(10):3205. doi: 10.3390/ijms19103205. Int J Mol Sci. 2018. PMID: 30336561 Free PMC article. Review.
References
-
- Plemper RK, Bohmler S, Bordallo J, Sommer T, Wolf DH.. Mutant analysis links the translocon and BiP to retrograde protein transport for ER degradation. Nature. 1997;388:891–5. - PubMed
-
- Brodsky JL, Werner ED, Dubas ME, Goeckeler JL, Kruse KB, McCracken AA.. The requirement for molecular chaperones during endoplasmic reticulum-associated protein degradation demonstrates that protein export and import are mechanistically distinct. J Biol Chem. 1999;274:3453–60. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous