

Autophagy and Obesity-Related Lung Disease
- PMID: 26900794
- PMCID: PMC5455357
- DOI: 10.1165/rcmb.2016-0045PS
Autophagy and Obesity-Related Lung Disease
Abstract
Obesity-related disease is a significant source of premature death and economic burden globally. It is also a common comorbidity in patients suffering from lung disease, affecting both severity and treatment success. However, this complex association between obesity and the lung is poorly understood. Autophagy is a self-recycling homeostatic process that has been linked to beneficial or deleterious effects, depending on the specific lung disease. Obesity affects autophagy in a tissue-specific manner, activating autophagy in adipocytes and impairing autophagy in hepatocytes, immune cells, and pancreatic β-cells, among others. Obesity is also characterized by chronic low-grade inflammation that can be modulated by the pro- and antiinflammatory effects of the autophagic machinery. Scant evidence exists regarding the impact of autophagy in obesity-related lung diseases, but there are communal pathways that could be related to disease pathogenesis. Important signaling molecules in obesity, including IL-17, leptin, adiponectin, NLRP3 inflammasome, and TLR-4, have been implicated in the pathogenesis of lung disease. These mediators are known to be modulated by autophagy activity. In this perspective, we highlight the recent advances in the understanding of autophagy in obesity-related conditions, as well as the potential mechanisms that can link autophagy and obesity in the pathogenesis of lung disease.
Keywords: autophagy; inflammation; lipid metabolism; lung disease; obesity.
Figures
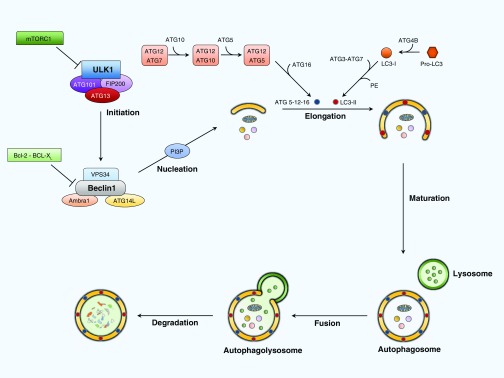
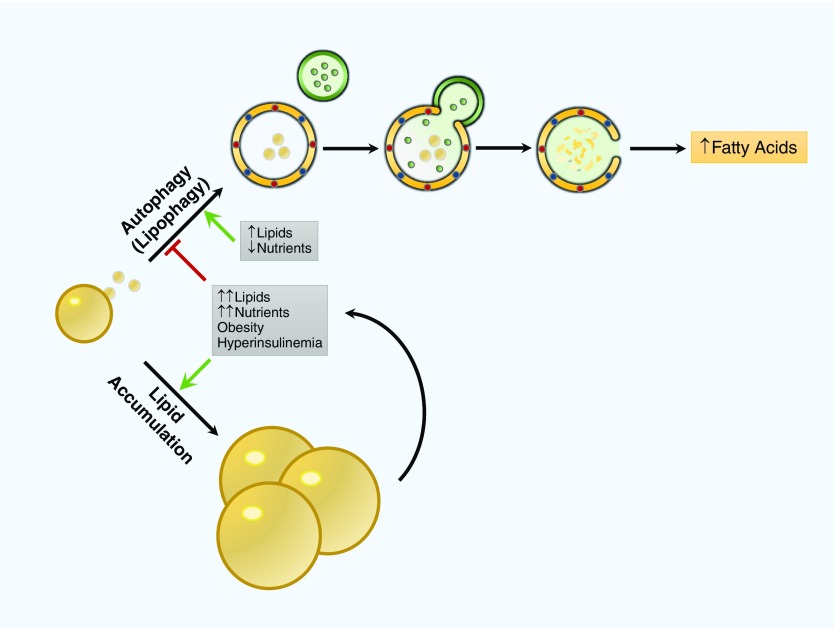
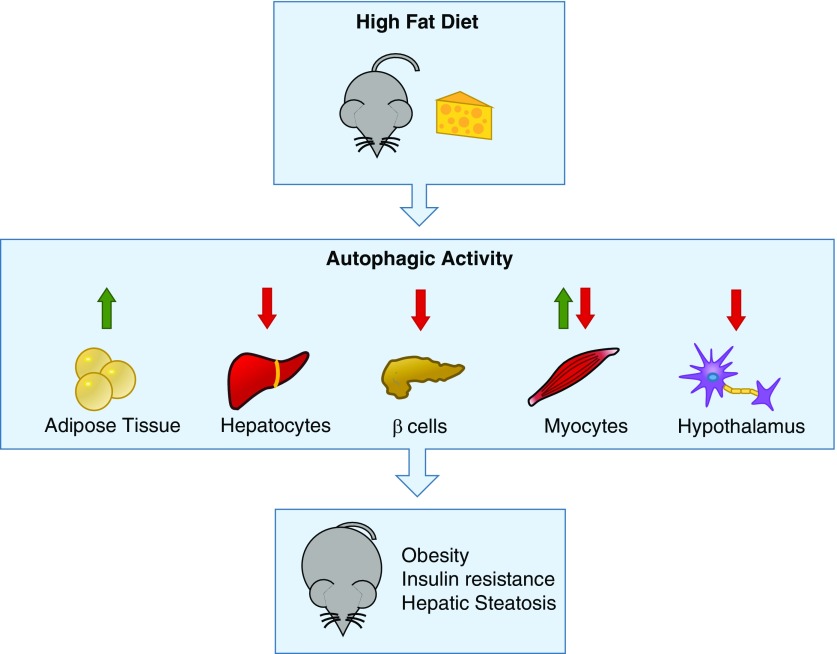
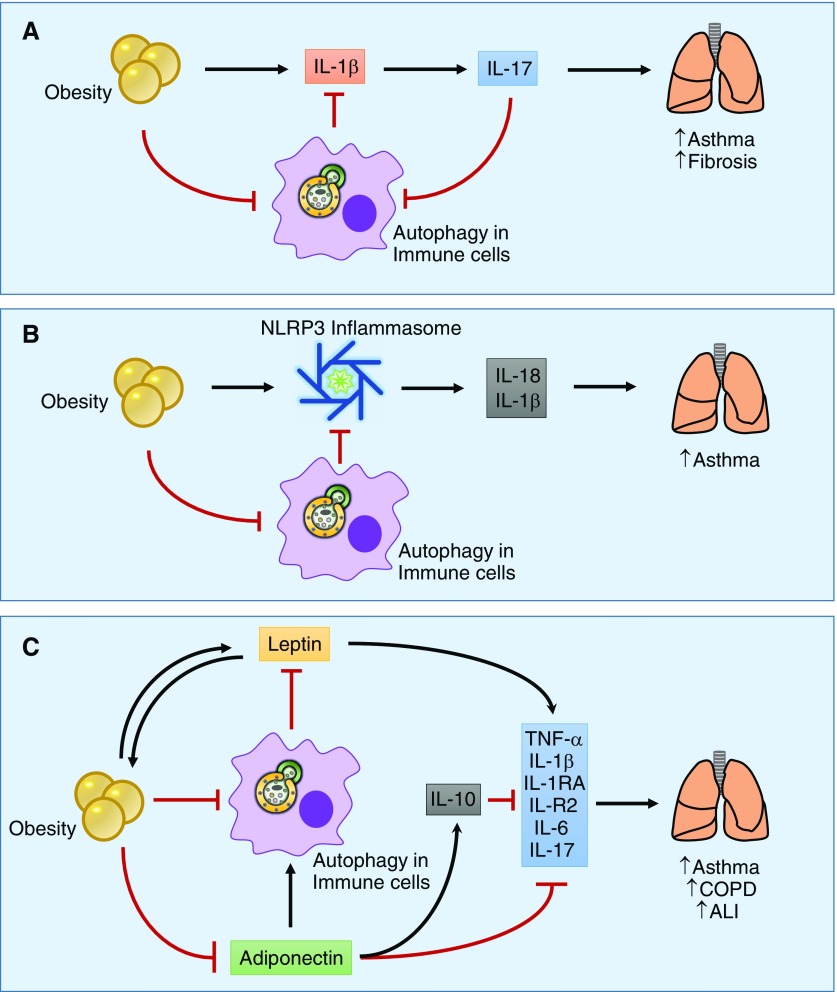
References
-
- World Health OrganizationObesity and overweight [Internet]. World Health Organization; 2015. [accessed 10 Jan 2016]Available from: http://www.who.int/mediacentre/factsheets/fs311/en/
-
- McNeil H, Segal L Quality of Life and Obesity. The Centre for Health Program Evaluation Monash University. 1999. Research report 17.
-
- Versini M, Jeandel PY, Rosenthal E, Shoenfeld Y. Obesity in autoimmune diseases: not a passive bystander. Autoimmun Rev. 2014;13:981–1000. - PubMed
-
- Karrowni W, Kennedy K, Jones P. Obesity paradox among survivors of acute myocardial infarction and its interaction with time. J Am Coll Cardiol. 2015;65(10S):A31.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
