

Integrated genomics and proteomics define huntingtin CAG length-dependent networks in mice
- PMID: 26900923
- PMCID: PMC5984042
- DOI: 10.1038/nn.4256
Integrated genomics and proteomics define huntingtin CAG length-dependent networks in mice
Abstract
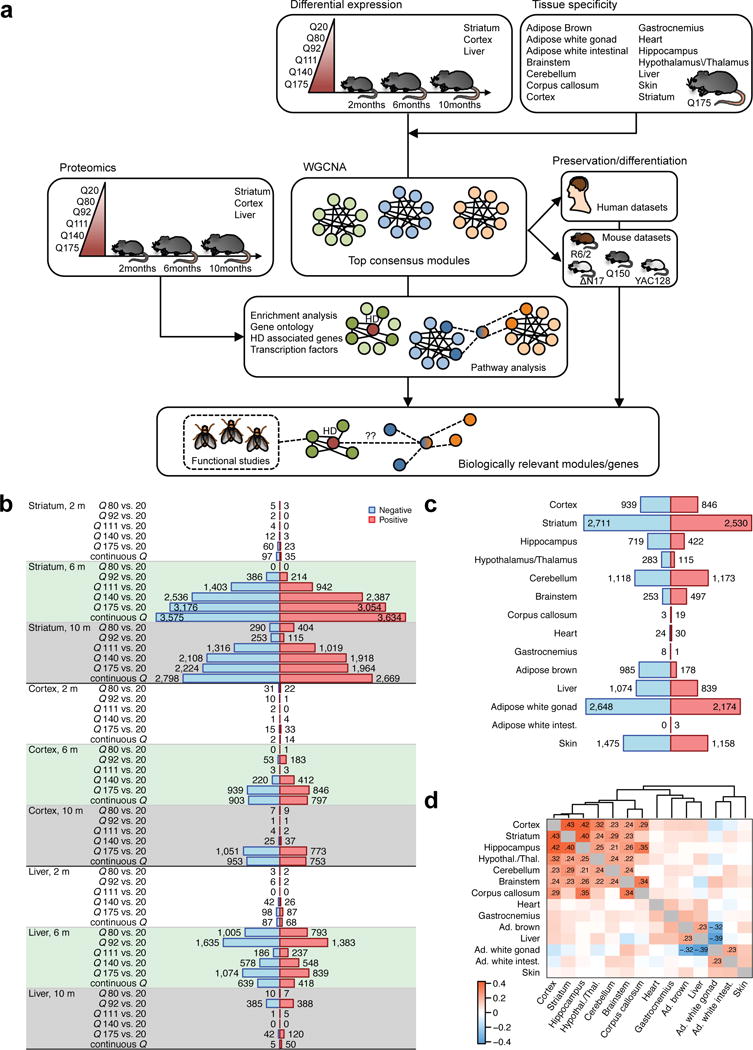
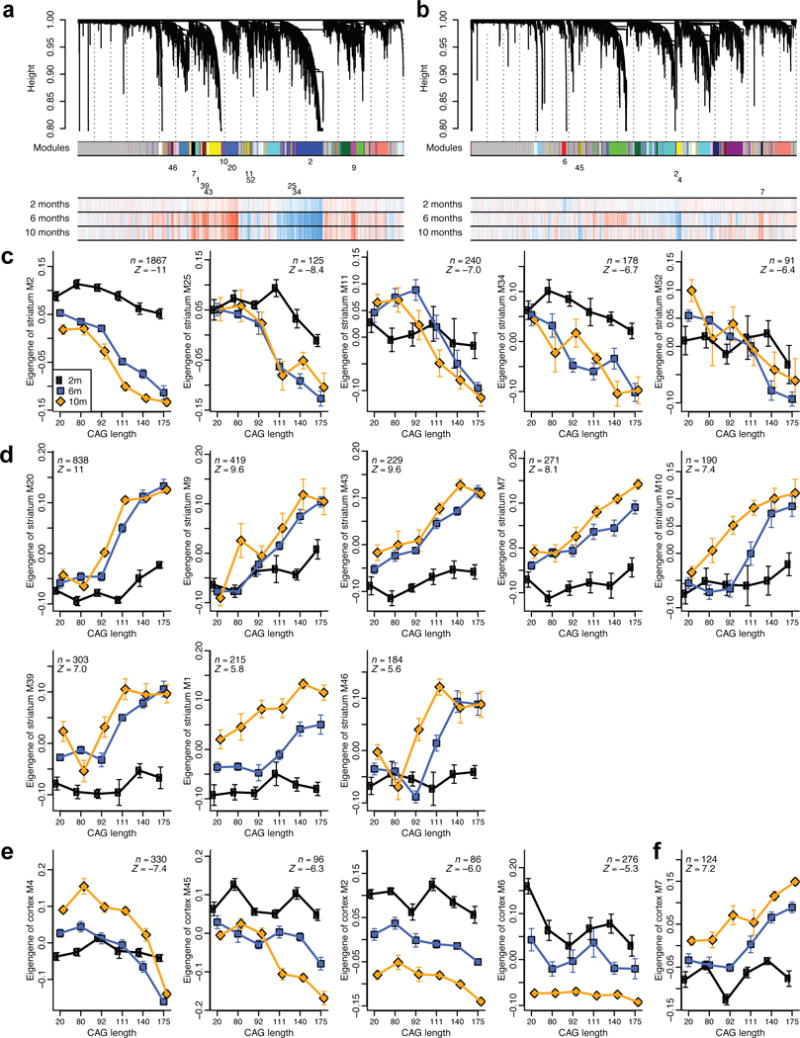
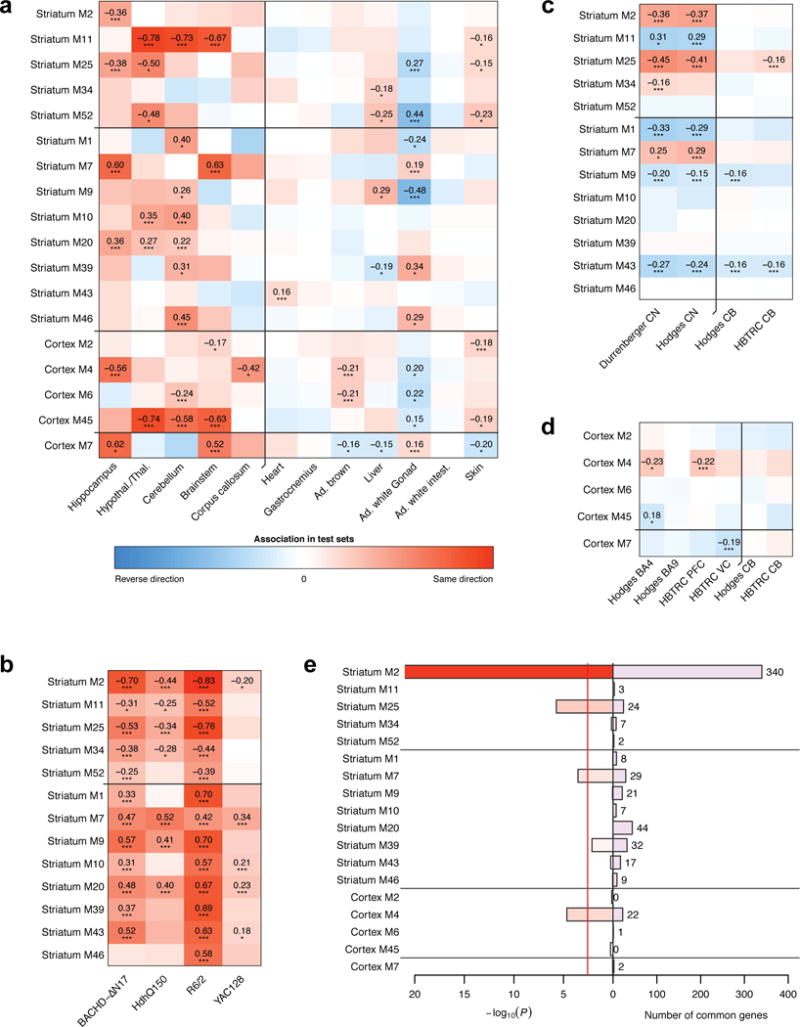
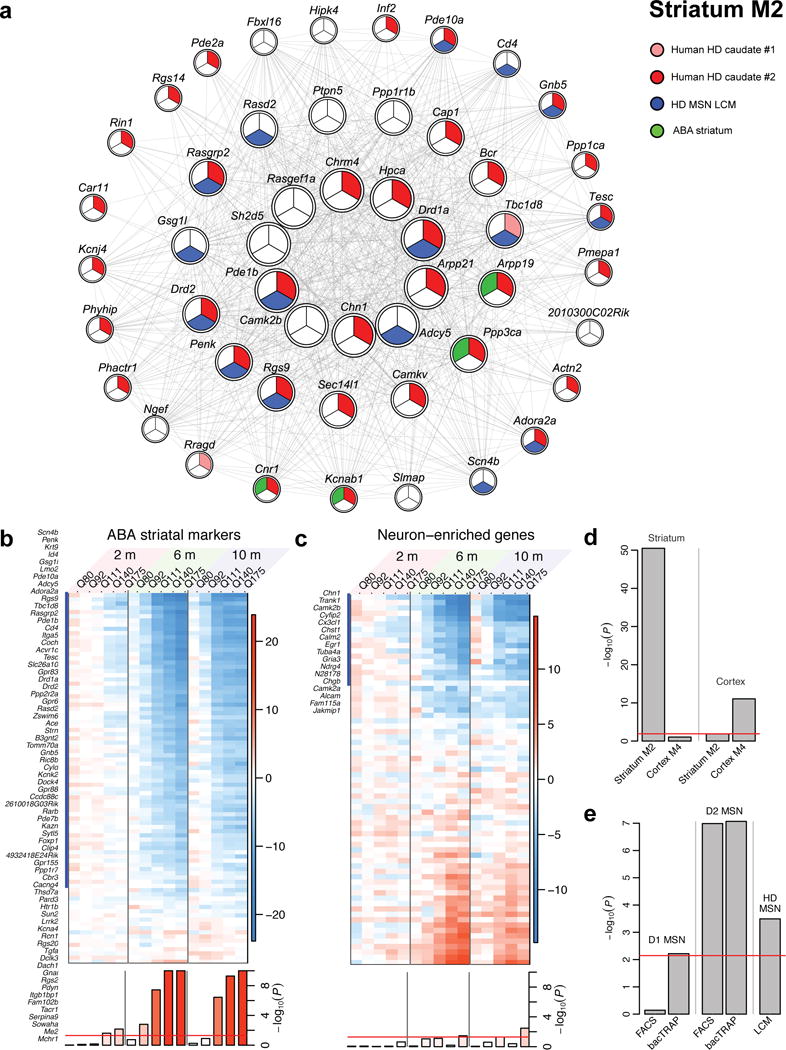
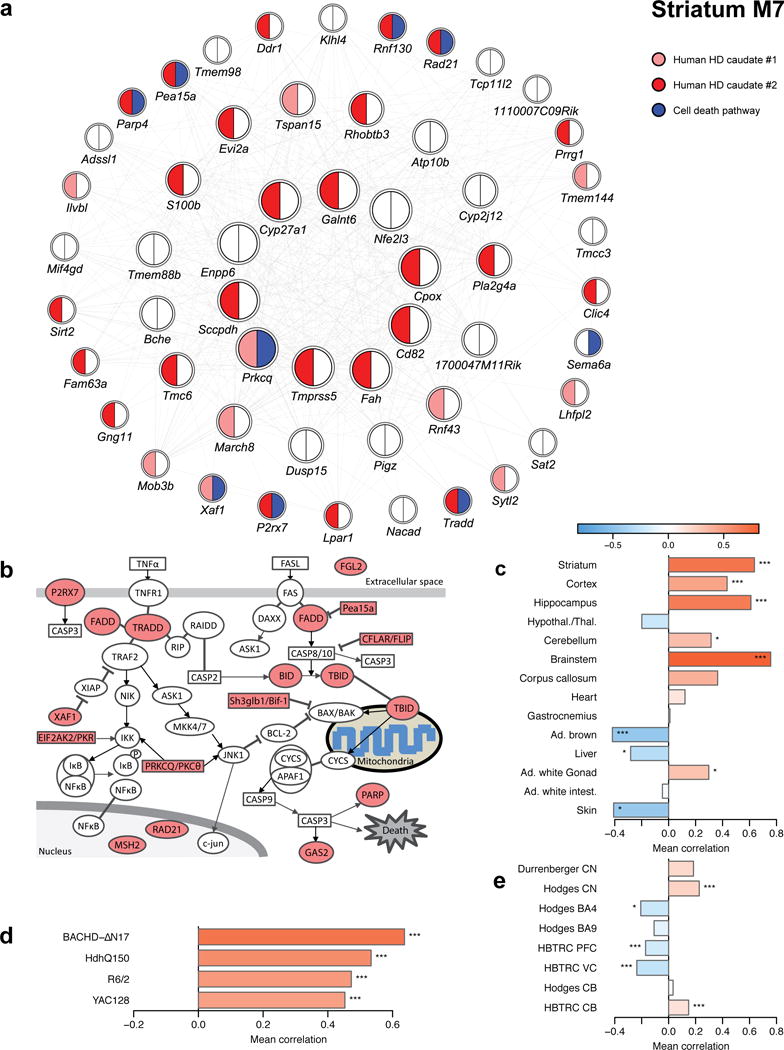
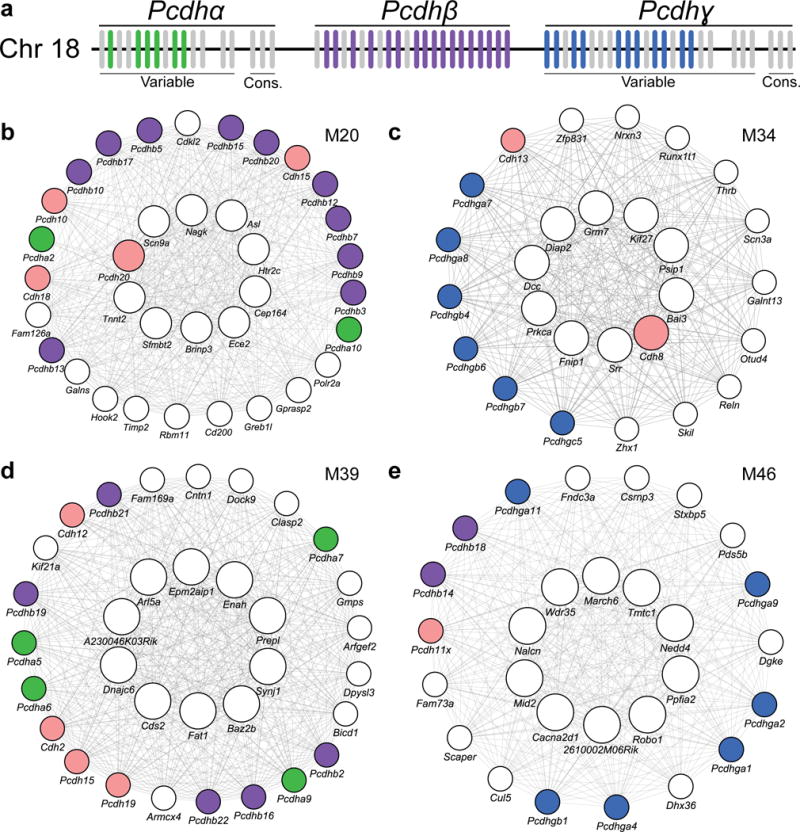
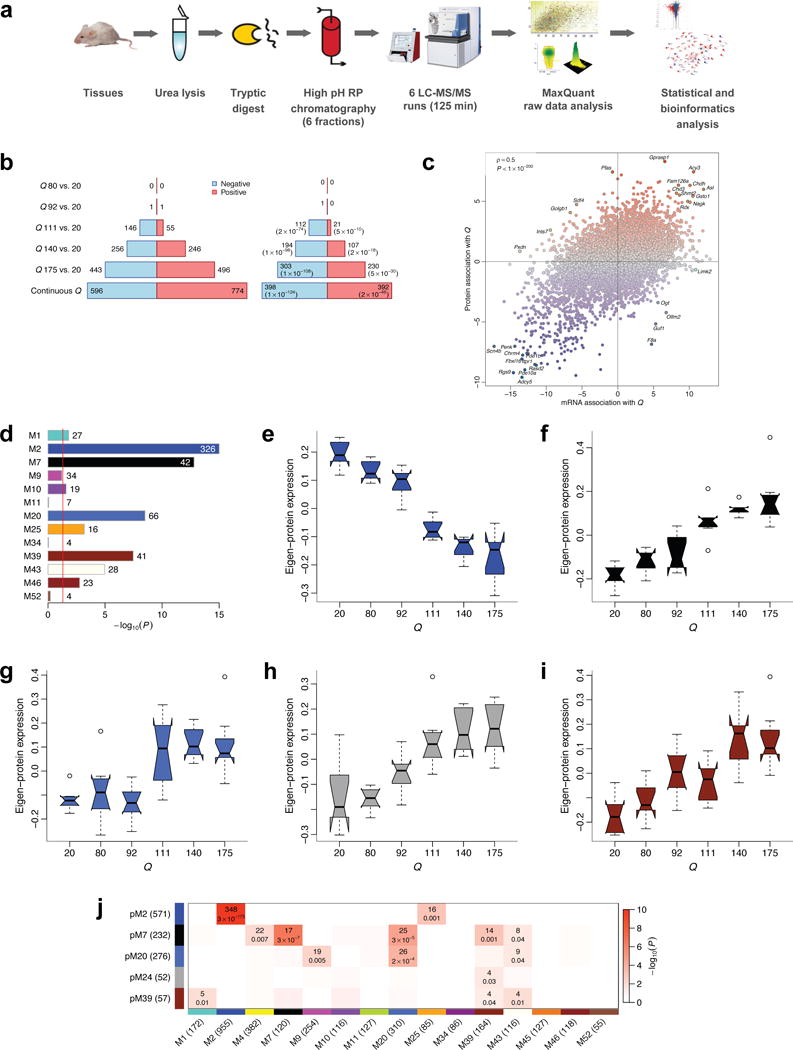
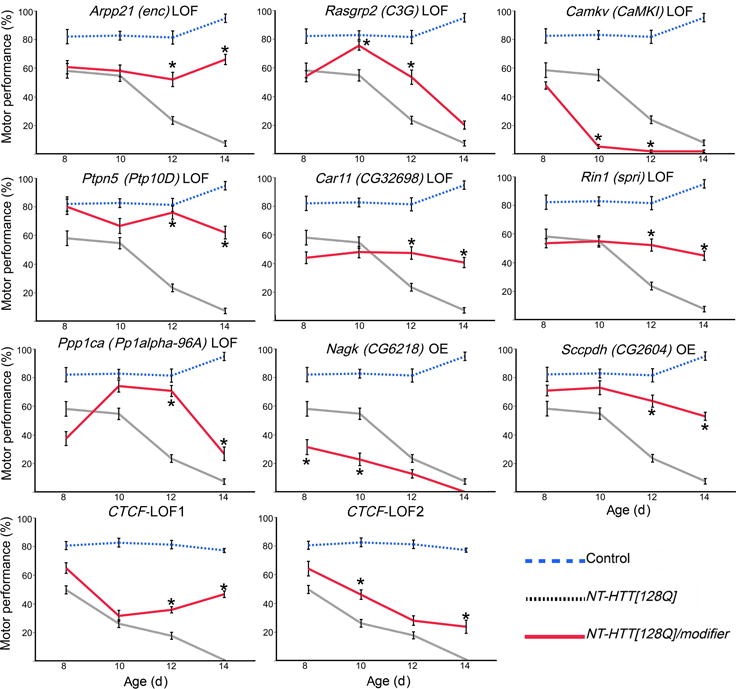
To gain insight into how mutant huntingtin (mHtt) CAG repeat length modifies Huntington's disease (HD) pathogenesis, we profiled mRNA in over 600 brain and peripheral tissue samples from HD knock-in mice with increasing CAG repeat lengths. We found repeat length-dependent transcriptional signatures to be prominent in the striatum, less so in cortex, and minimal in the liver. Coexpression network analyses revealed 13 striatal and 5 cortical modules that correlated highly with CAG length and age, and that were preserved in HD models and sometimes in patients. Top striatal modules implicated mHtt CAG length and age in graded impairment in the expression of identity genes for striatal medium spiny neurons and in dysregulation of cyclic AMP signaling, cell death and protocadherin genes. We used proteomics to confirm 790 genes and 5 striatal modules with CAG length-dependent dysregulation at the protein level, and validated 22 striatal module genes as modifiers of mHtt toxicities in vivo.
Conflict of interest statement
The authors declare that they have no competing interests that might be perceived to influence the results and/or discussion reported in this article.
Figures
References
-
- Ross CA, et al. Huntington disease: natural history, biomarkers and prospects for therapeutics. Nature reviews. Neurology. 2014;10:204–216. - PubMed
-
- Vonsattel JP, DiFiglia M. Huntington disease. Journal of neuropathology and experimental neurology. 1998;57:369–384. - PubMed
-
- A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell. 1993;72:971–983. - PubMed
-
- Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annual review of neuroscience. 2007;30:575–621. - PubMed
-
- Gusella JF, MacDonald ME. Molecular genetics: unmasking polyglutamine triggers in neurodegenerative disease. Nature reviews. Neuroscience. 2000;1:109–115. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
