

Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas
- PMID: 26902283
- PMCID: PMC4783472
- DOI: 10.1038/cr.2016.23
Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas
Abstract
Single-cell genome, DNA methylome, and transcriptome sequencing methods have been separately developed. However, to accurately analyze the mechanism by which transcriptome, genome and DNA methylome regulate each other, these omic methods need to be performed in the same single cell. Here we demonstrate a single-cell triple omics sequencing technique, scTrio-seq, that can be used to simultaneously analyze the genomic copy-number variations (CNVs), DNA methylome, and transcriptome of an individual mammalian cell. We show that large-scale CNVs cause proportional changes in RNA expression of genes within the gained or lost genomic regions, whereas these CNVs generally do not affect DNA methylation in these regions. Furthermore, we applied scTrio-seq to 25 single cancer cells derived from a human hepatocellular carcinoma tissue sample. We identified two subpopulations within these cells based on CNVs, DNA methylome, or transcriptome of individual cells. Our work offers a new avenue of dissecting the complex contribution of genomic and epigenomic heterogeneities to the transcriptomic heterogeneity within a population of cells.
Figures
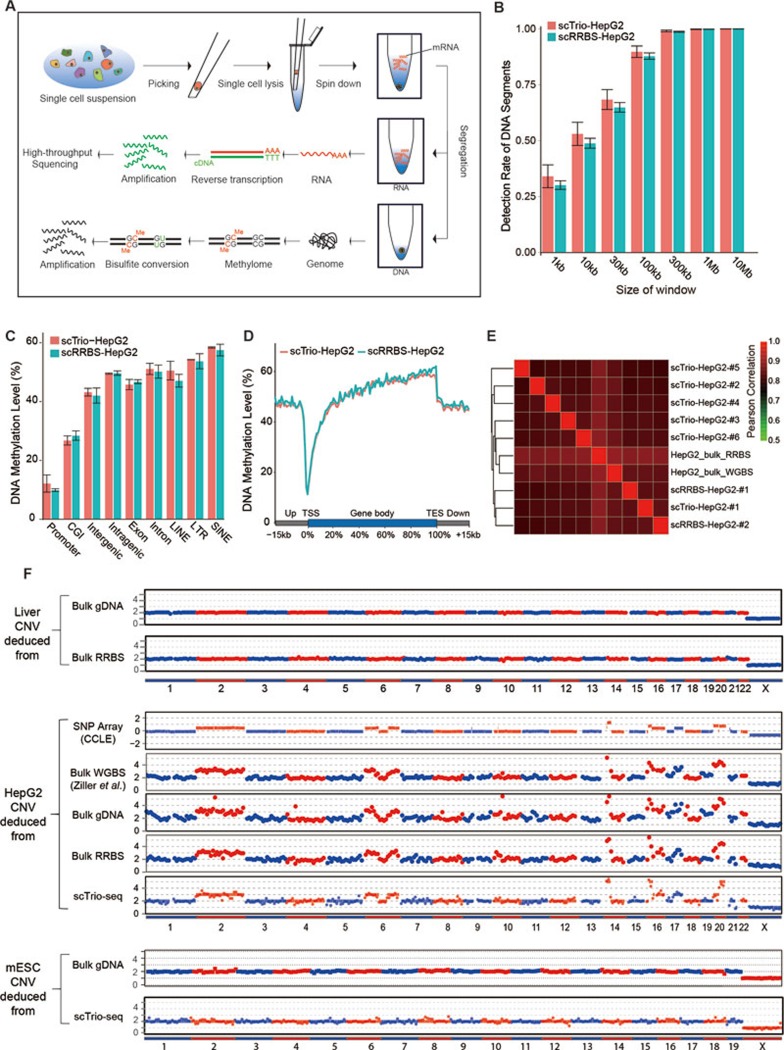
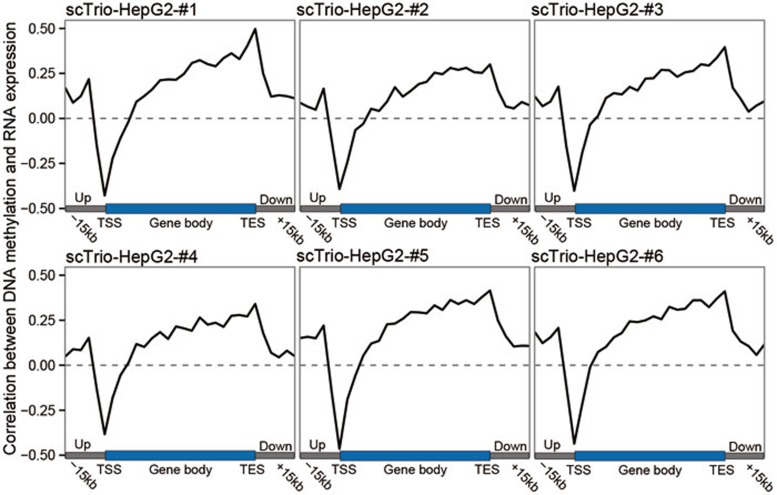
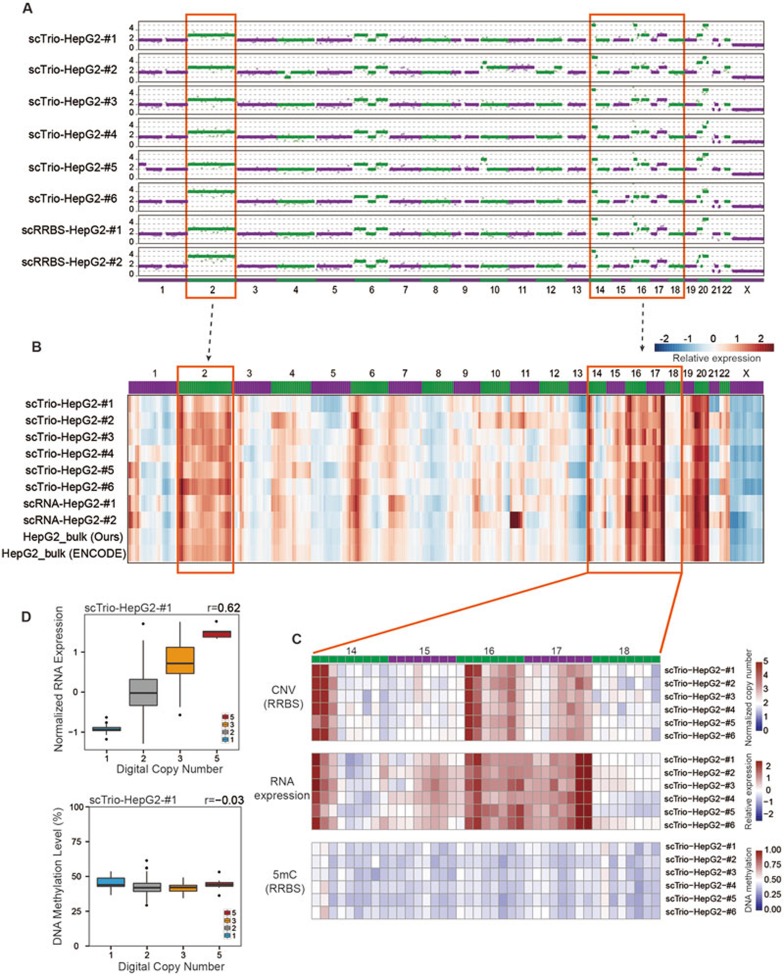
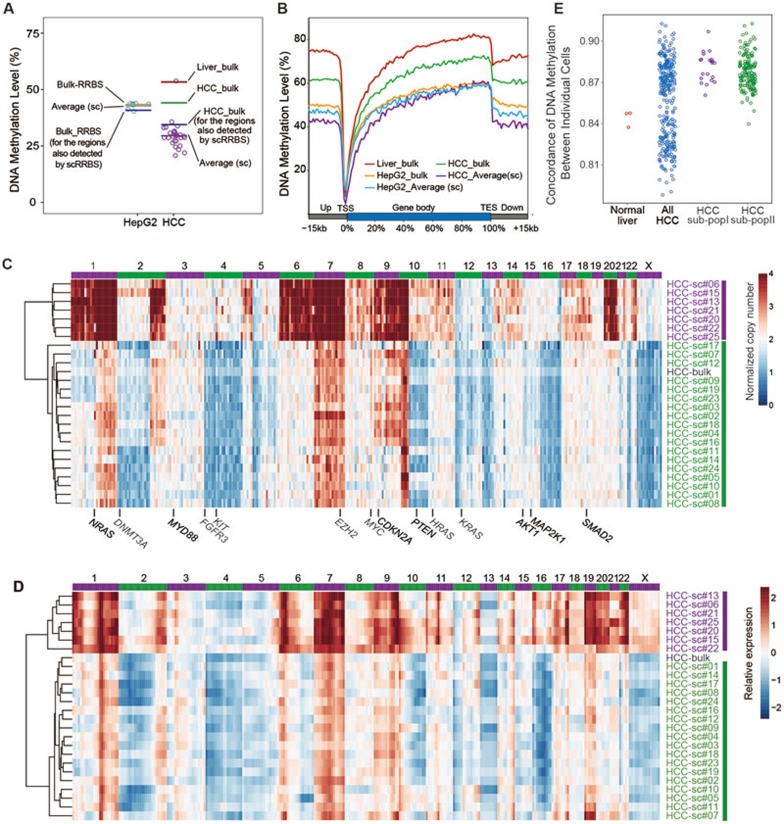
References
-
- Wen L, Tang F. Reconstructing complex tissues from single-cell analyses. Cell 2014; 157:771–773. - PubMed
-
- Shapiro E, Biezuner T, Linnarsson S. Single-cell sequencing-based technologies will revolutionize whole-organism science. Nat Rev Genet 2013; 14:618–630. - PubMed
-
- Hashimshony T, Wagner F, Sher N, Yanai I. CEL-Seq: single-cell RNA-Seq by multiplexed linear amplification. Cell Rep 2012; 2:666–673. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
