

From Drug Screening to Target Deconvolution: a Target-Based Drug Discovery Pipeline Using Leishmania Casein Kinase 1 Isoform 2 To Identify Compounds with Antileishmanial Activity
- PMID: 26902771
- PMCID: PMC4862455
- DOI: 10.1128/AAC.00021-16
From Drug Screening to Target Deconvolution: a Target-Based Drug Discovery Pipeline Using Leishmania Casein Kinase 1 Isoform 2 To Identify Compounds with Antileishmanial Activity
Abstract
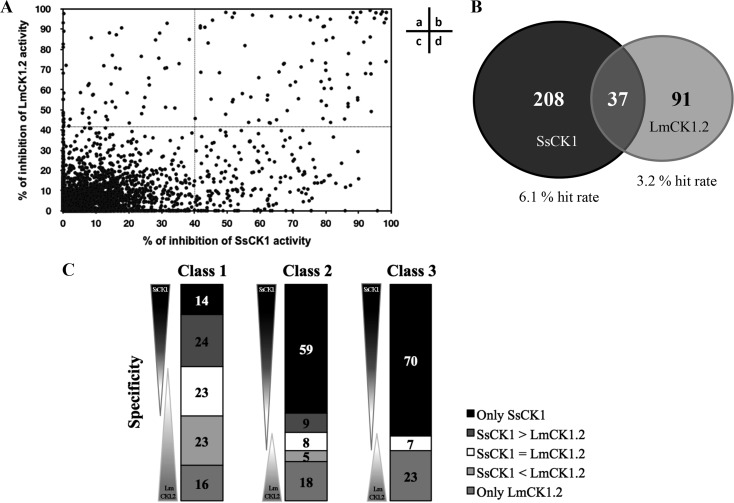
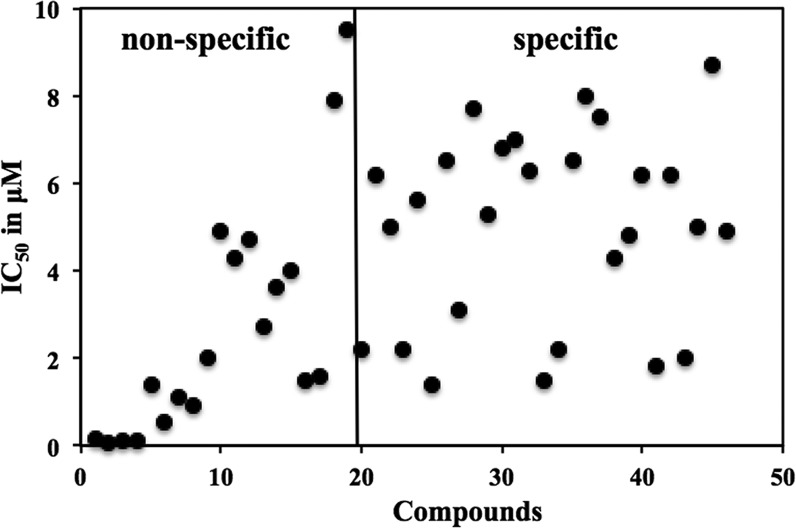
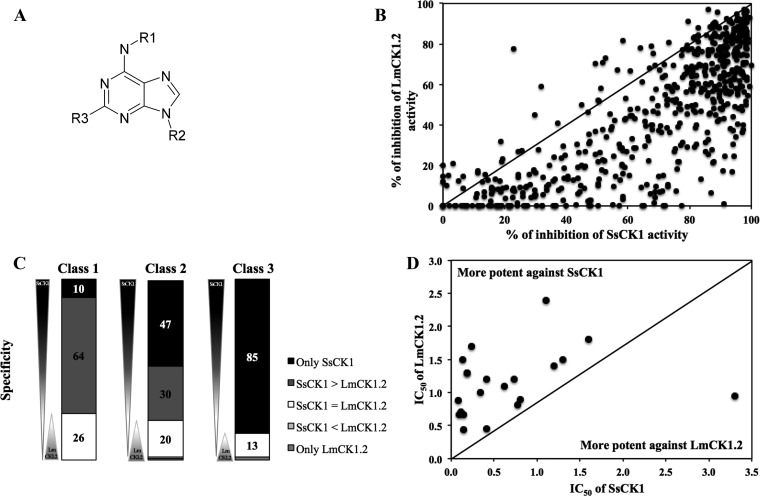
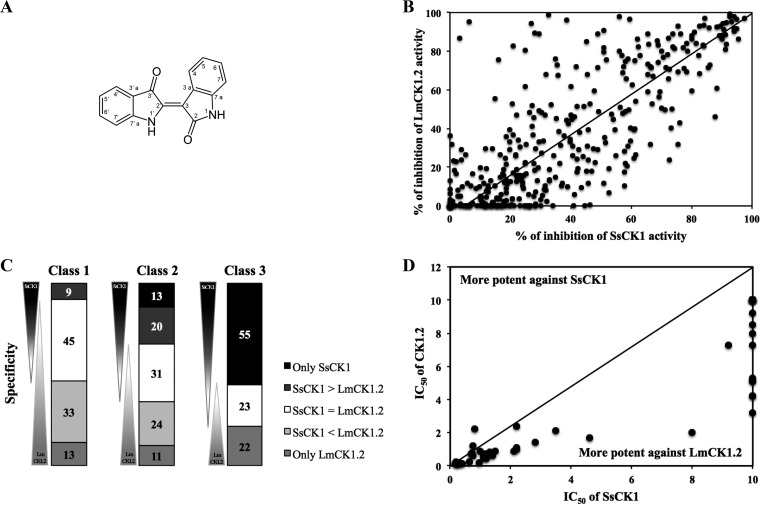
Existing therapies for leishmaniases present significant limitations, such as toxic side effects, and are rendered inefficient by parasite resistance. It is of utmost importance to develop novel drugs targeting Leishmania that take these two limitations into consideration. We thus chose a target-based approach using an exoprotein kinase, Leishmania casein kinase 1.2 (LmCK1.2) that was recently shown to be essential for intracellular parasite survival and infectivity. We developed a four-step pipeline to identify novel selective antileishmanial compounds. In step 1, we screened 5,018 compounds from kinase-biased libraries with Leishmania and mammalian CK1 in order to identify hit compounds and assess their specificity. For step 2, we selected 88 compounds among those with the lowest 50% inhibitory concentration to test their biological activity on host-free parasites using a resazurin reduction assay and on intramacrophagic amastigotes using a high content phenotypic assay. Only 75 compounds showed antileishmanial activity and were retained for step 3 to evaluate their toxicity against mouse macrophages and human cell lines. The four compounds that displayed a selectivity index above 10 were then assessed for their affinity to LmCK1.2 using a target deconvolution strategy in step 4. Finally, we retained two compounds, PP2 and compound 42, for which LmCK1.2 seems to be the primary target. Using this four-step pipeline, we identify from several thousand molecules, two lead compounds with a selective antileishmanial activity.
Copyright © 2016, American Society for Microbiology. All Rights Reserved.
Figures
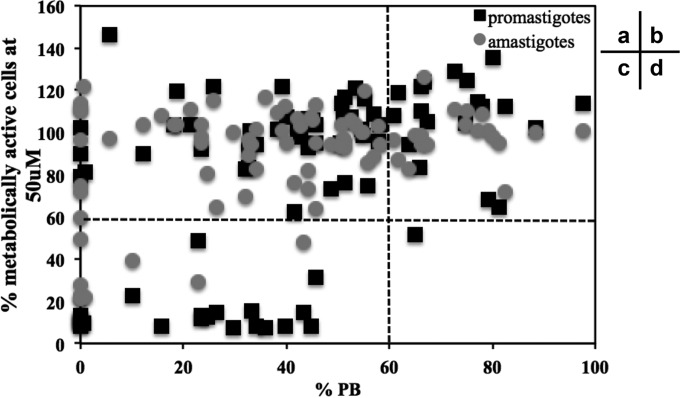
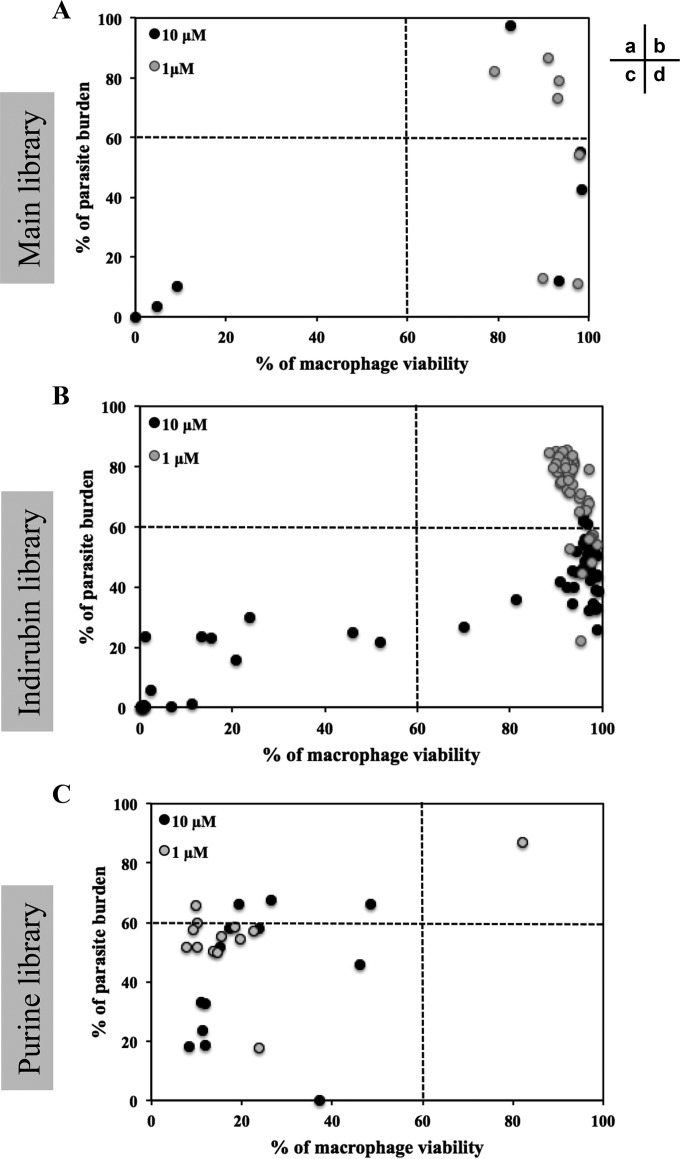
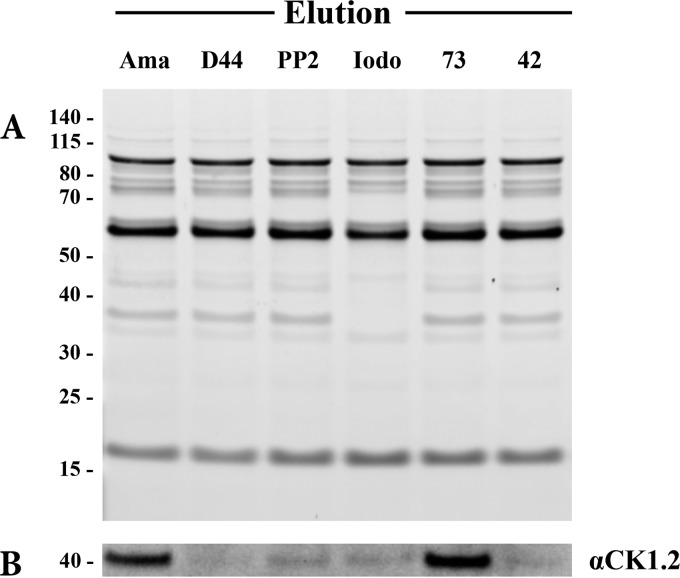
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
