

Trimethylamine N-Oxide Promotes Vascular Inflammation Through Signaling of Mitogen-Activated Protein Kinase and Nuclear Factor-κB
- PMID: 26903003
- PMCID: PMC4802459
- DOI: 10.1161/JAHA.115.002767
Trimethylamine N-Oxide Promotes Vascular Inflammation Through Signaling of Mitogen-Activated Protein Kinase and Nuclear Factor-κB
Abstract
Background: The choline-derived metabolite trimethylamine N-oxide (TMAO) has been demonstrated to contribute to atherosclerosis and is associated with coronary artery disease risk.
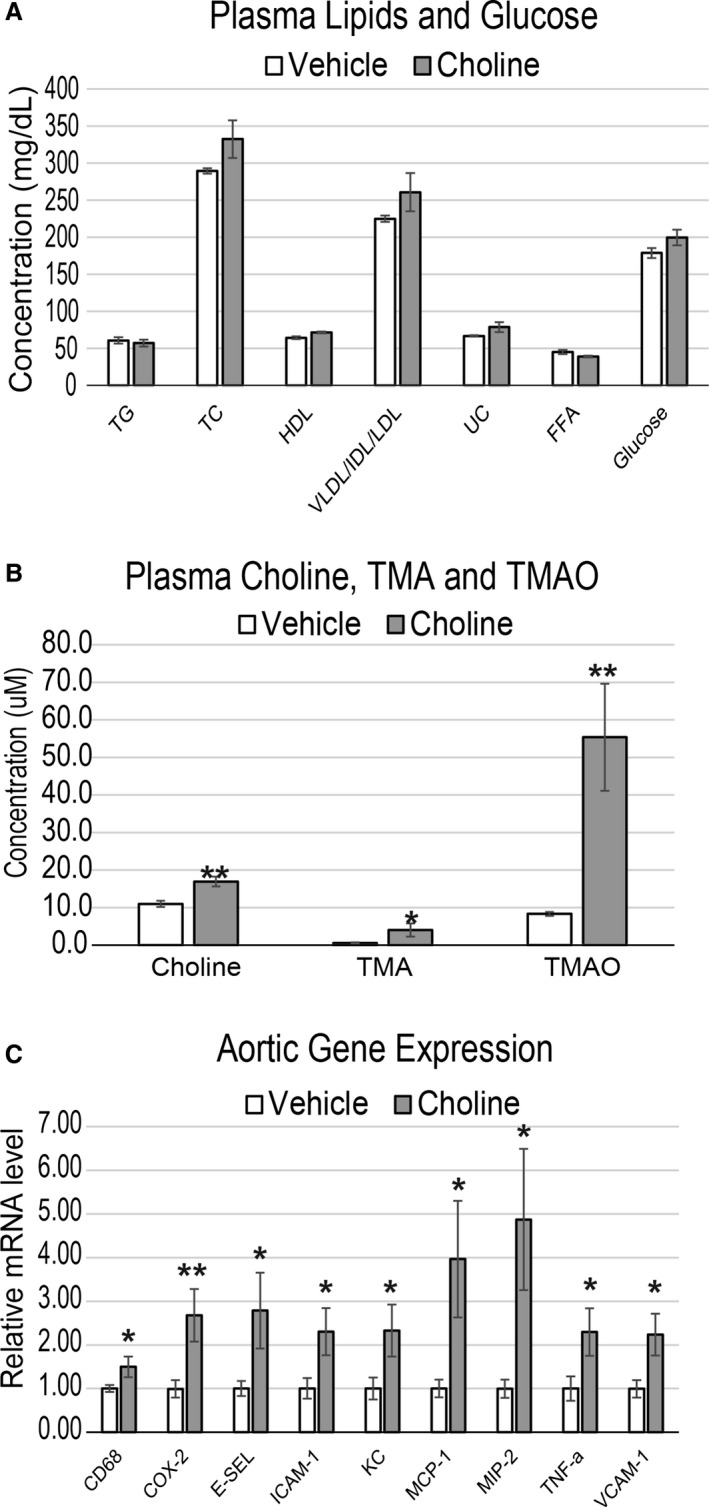
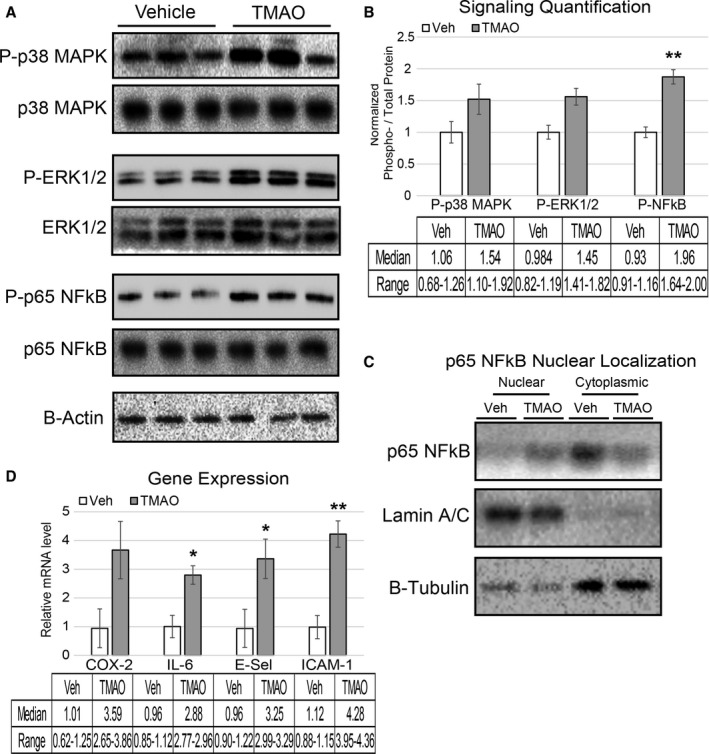
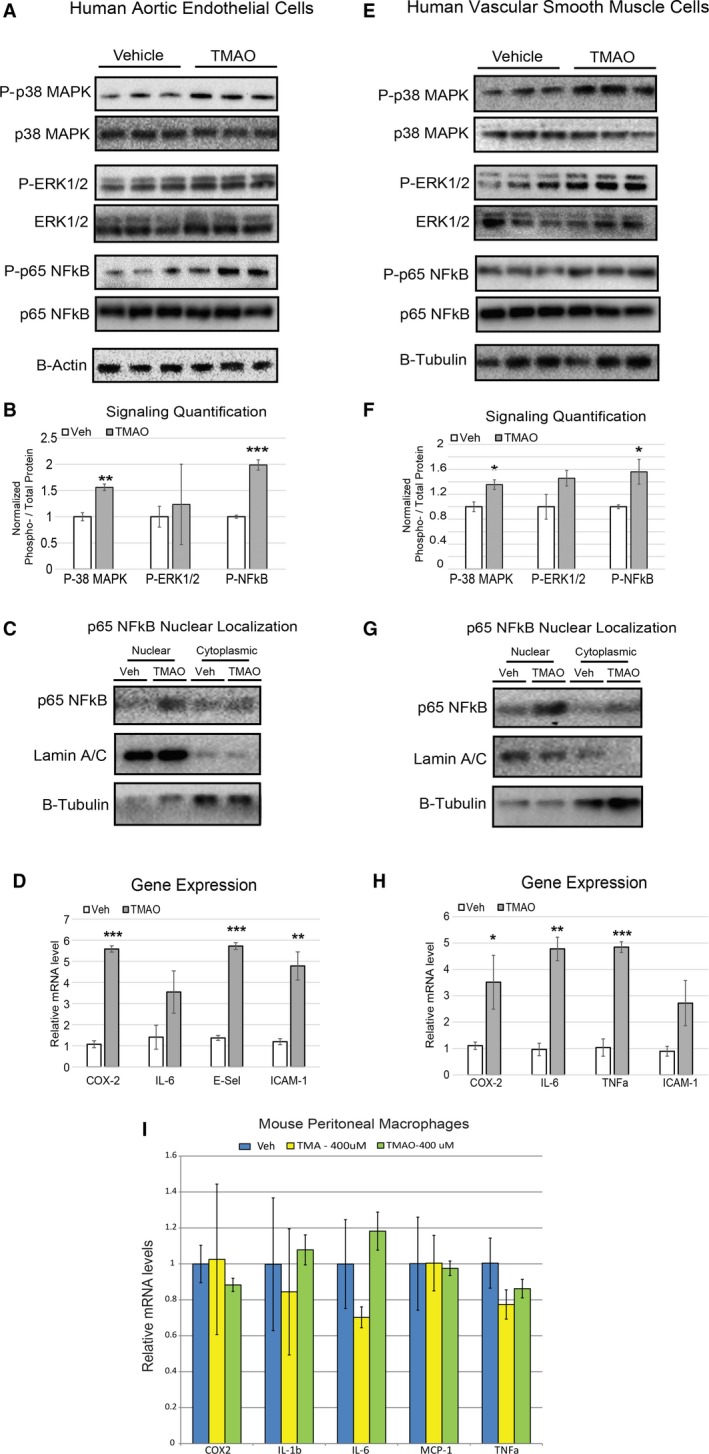
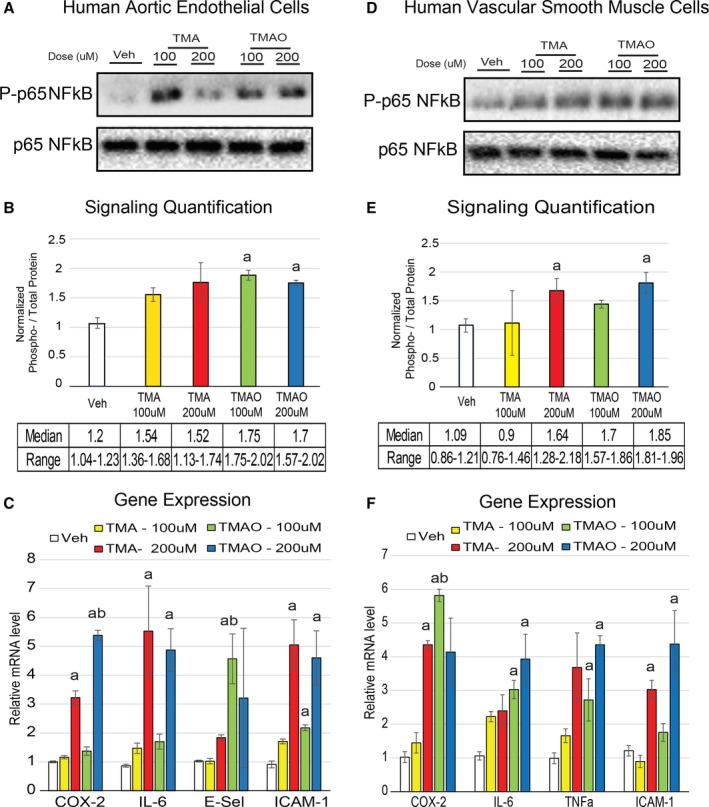
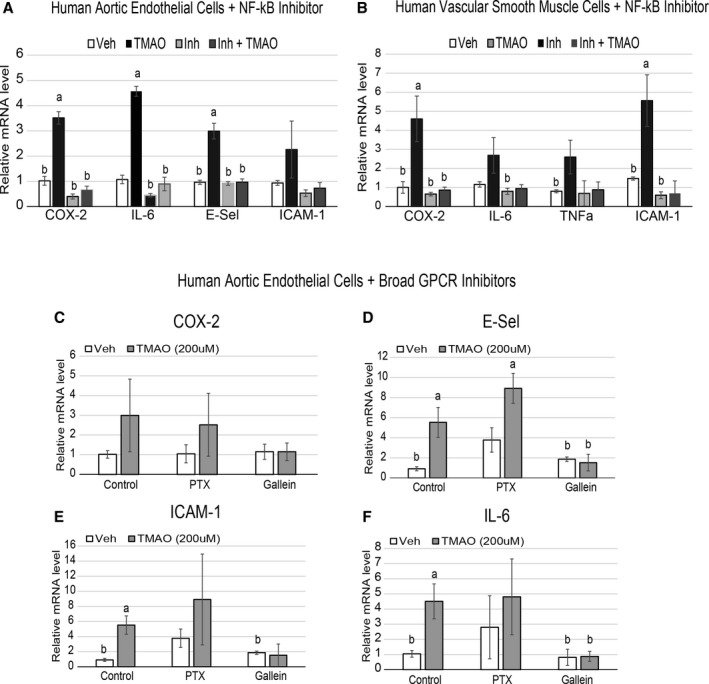
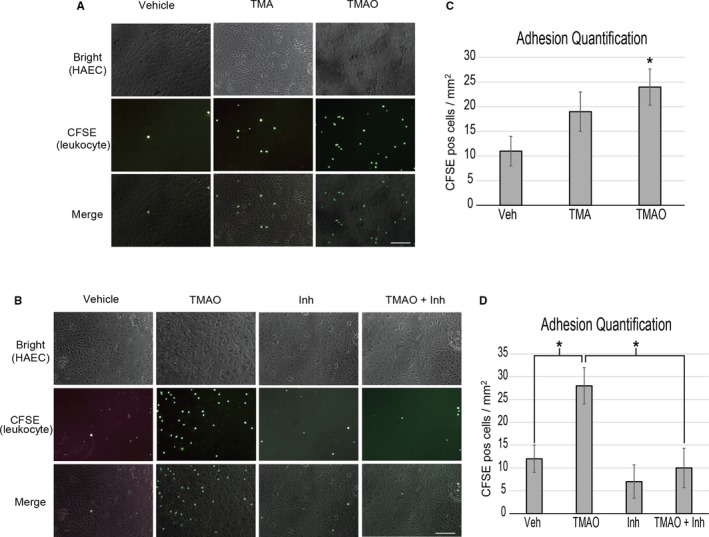
Methods and results: We explored the impact of TMAO on endothelial and smooth muscle cell function in vivo, focusing on disease-relevant outcomes for atherogenesis. Initially, we observed that aortas of LDLR(-/-) mice fed a choline diet showed elevated inflammatory gene expression compared with controls. Acute TMAO injection at physiological levels was sufficient to induce the same inflammatory markers and activate the well-known mitogen-activated protein kinase, extracellular signal-related kinase, and nuclear factor-κB signaling cascade. These observations were recapitulated in primary human aortic endothelial cells and vascular smooth muscle cells. We also found that TMAO promotes recruitment of activated leukocytes to endothelial cells. Through pharmacological inhibition, we further showed that activation of nuclear factor-κB signaling was necessary for TMAO to induce inflammatory gene expression in both of these relevant cell types as well as endothelial cell adhesion of leukocytes.
Conclusions: Our results suggest a likely contributory mechanism for TMAO-dependent enhancement in atherosclerosis and cardiovascular risks.
Keywords: atherosclerosis; cardiovascular disease; endothelial cell; inflammation; leukocyte adhesion; nuclear factor‐κB signaling; trimethylamine N‐oxide; vascular smooth muscle cell.
© 2016 The Authors. Published on behalf of the American Heart Association, Inc., by Wiley Blackwell.
Figures
References
-
- Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, Britt EB, Fu X, Wu Y, Li L, Smith JD, DiDonato JA, Chen J, Li H, Wu GD, Lewis JD, Warrier M, Brown JM, Krauss RM, Tang WH, Bushman FD, Lusis AJ, Hazen SL. Intestinal microbiota metabolism of L‐carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. 2013;19:576–585. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
