

Krüppeling erythropoiesis: an unexpected broad spectrum of human red blood cell disorders due to KLF1 variants
- PMID: 26903544
- PMCID: PMC4832505
- DOI: 10.1182/blood-2016-01-694331
Krüppeling erythropoiesis: an unexpected broad spectrum of human red blood cell disorders due to KLF1 variants
Abstract
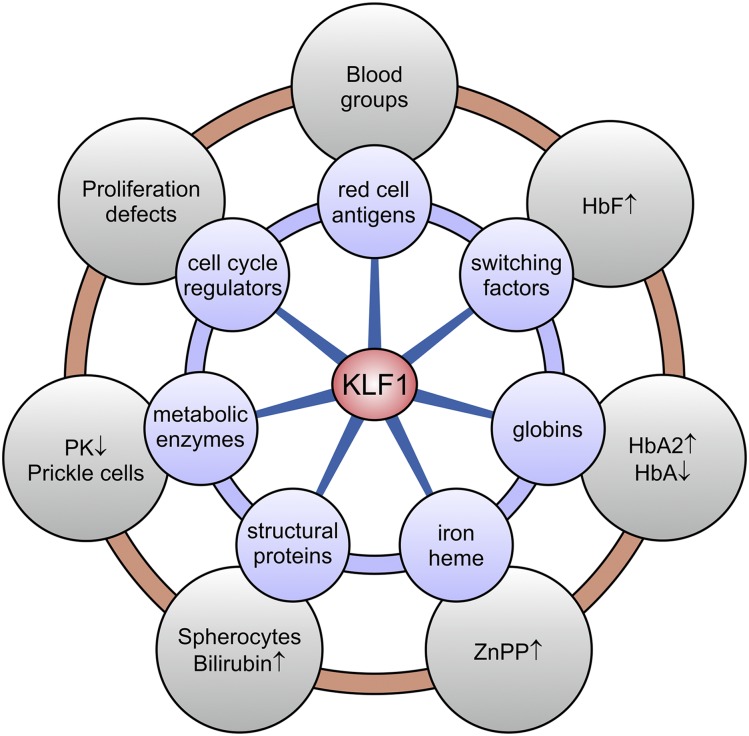
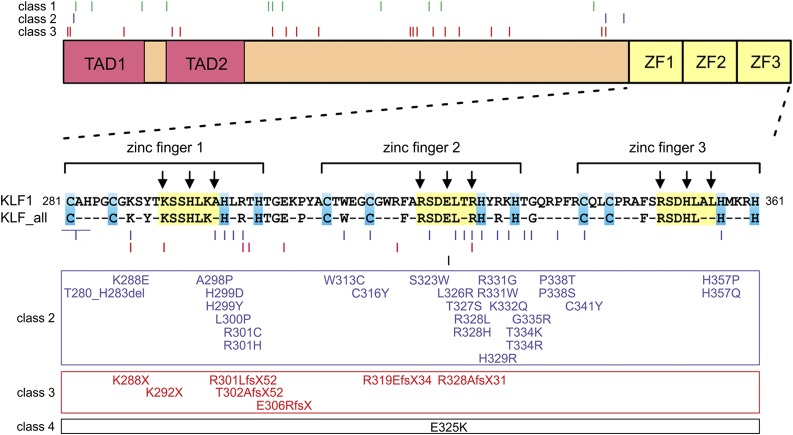
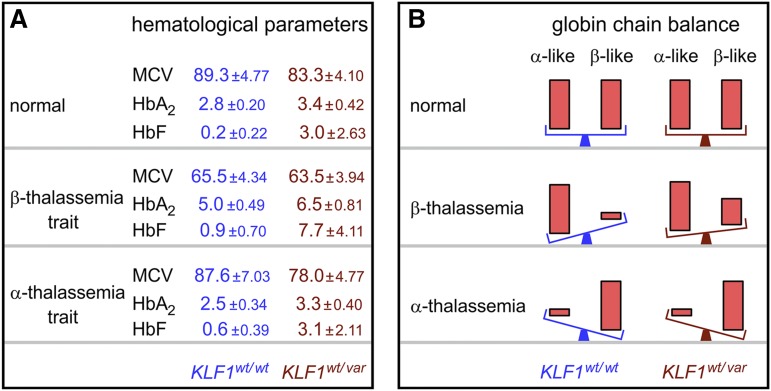
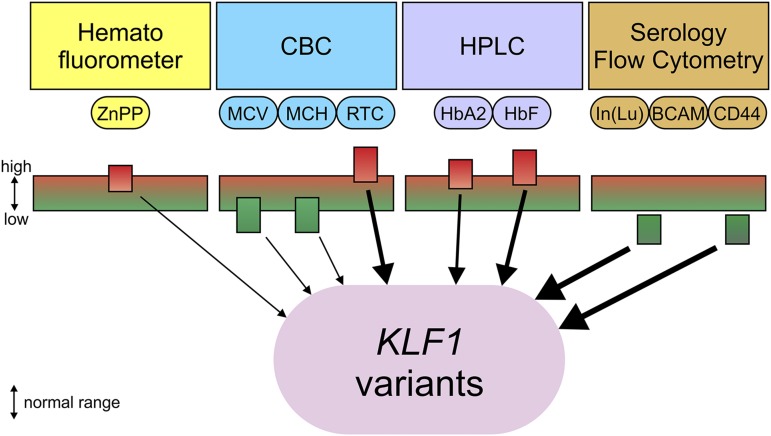
Until recently our approach to analyzing human genetic diseases has been to accurately phenotype patients and sequence the genes known to be associated with those phenotypes; for example, in thalassemia, the globin loci are analyzed. Sequencing has become increasingly accessible, and thus a larger panel of genes can be analyzed and whole exome and/or whole genome sequencing can be used when no variants are found in the candidate genes. By using such approaches in patients with unexplained anemias, we have discovered that a broad range of hitherto unrelated human red cell disorders are caused by variants in KLF1, a master regulator of erythropoiesis, which were previously considered to be extremely rare causes of human genetic disease.
© 2016 by The American Society of Hematology.
Figures
Similar articles
-
KLF1-null neonates display hydrops fetalis and a deranged erythroid transcriptome.Blood. 2015 Apr 9;125(15):2405-17. doi: 10.1182/blood-2014-08-590968. Epub 2015 Feb 27. Blood. 2015. PMID: 25724378 Free PMC article.
-
Differential role of Kruppel like factor 1 (KLF1) gene in red blood cell disorders.Genomics. 2019 Dec;111(6):1771-1776. doi: 10.1016/j.ygeno.2018.11.032. Epub 2018 Dec 5. Genomics. 2019. PMID: 30529538
-
Krüppel-Like Factor 1: A Pivotal Gene Regulator in Erythropoiesis.Cells. 2022 Sep 29;11(19):3069. doi: 10.3390/cells11193069. Cells. 2022. PMID: 36231031 Free PMC article. Review.
-
Kruppel-like factor 1 (KLF1), KLF2, and Myc control a regulatory network essential for embryonic erythropoiesis.Mol Cell Biol. 2012 Jul;32(13):2628-44. doi: 10.1128/MCB.00104-12. Epub 2012 May 7. Mol Cell Biol. 2012. PMID: 22566683 Free PMC article.
-
Impact of transcription factors KLF1 and GATA1 on red blood cell antigen expression: a review.Immunohematology. 2024 May 13;40(1):1-9. doi: 10.2478/immunohematology-2024-002. eCollection 2024 Apr 1. Immunohematology. 2024. PMID: 38739025 Review.
Cited by
-
Thalassemia and erythroid transcription factor KLF1 mutations associated with borderline hemoglobin A2 in the Thai population.Arch Med Sci. 2020 Aug 11;18(1):112-120. doi: 10.5114/aoms.2020.93392. eCollection 2022. Arch Med Sci. 2020. PMID: 35154532 Free PMC article.
-
Survey and evaluation of mutations in the human KLF1 transcription unit.Sci Rep. 2018 Apr 26;8(1):6587. doi: 10.1038/s41598-018-24962-3. Sci Rep. 2018. PMID: 29700354 Free PMC article.
-
Hematopoietic cell transplantation for congenital dyserythropoietic anemia IV caused by compound heterozygous KLF1 mutations.Ann Hematol. 2023 Jun;102(6):1621-1624. doi: 10.1007/s00277-023-05175-9. Epub 2023 Mar 31. Ann Hematol. 2023. PMID: 37002443 No abstract available.
-
Severe anemia caused by dominant mutations in Krüppel-like factor 1 (KLF1).Mutat Res Rev Mutat Res. 2020 Oct-Dec;786:108336. doi: 10.1016/j.mrrev.2020.108336. Epub 2020 Oct 9. Mutat Res Rev Mutat Res. 2020. PMID: 33339573 Free PMC article. Review.
-
Sickle Cell Anemia and Its Phenotypes.Annu Rev Genomics Hum Genet. 2018 Aug 31;19:113-147. doi: 10.1146/annurev-genom-083117-021320. Epub 2018 Apr 11. Annu Rev Genomics Hum Genet. 2018. PMID: 29641911 Free PMC article. Review.
References
-
- Singleton BK, Burton NM, Green C, Brady RL, Anstee DJ. Mutations in EKLF/KLF1 form the molecular basis of the rare blood group In(Lu) phenotype. Blood. 2008;112(5):2081–2088. - PubMed
-
- Viprakasit V, Ekwattanakit S, Riolueang S, et al. Mutations in Kruppel-like factor 1 cause transfusion-dependent hemolytic anemia and persistence of embryonic globin gene expression. Blood. 2014;123(10):1586–1595. - PubMed
-
- Jaffray JA, Mitchell WB, Gnanapragasam MN, et al. Erythroid transcription factor EKLF/KLF1 mutation causing congenital dyserythropoietic anemia type IV in a patient of Taiwanese origin: review of all reported cases and development of a clinical diagnostic paradigm. Blood Cells Mol Dis. 2013;51(2):71–75. - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
