

Advancing clinical development pathways for new CFTR modulators in cystic fibrosis
- PMID: 26903594
- PMCID: PMC4853537
- DOI: 10.1136/thoraxjnl-2015-208123
Advancing clinical development pathways for new CFTR modulators in cystic fibrosis
Abstract
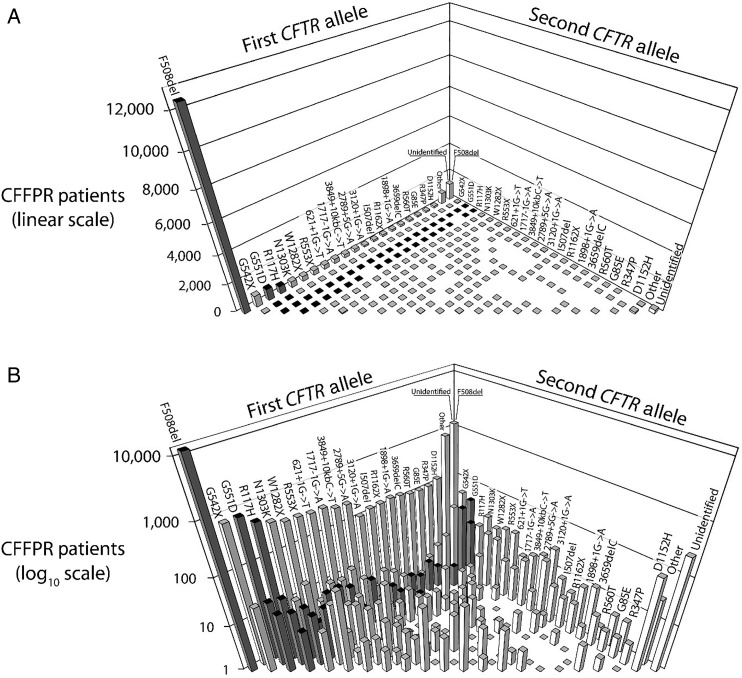
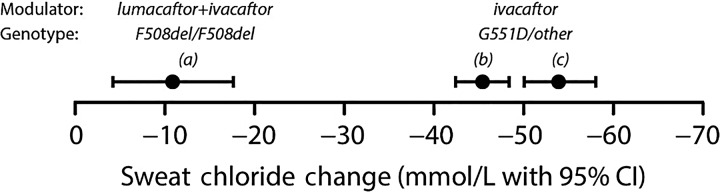
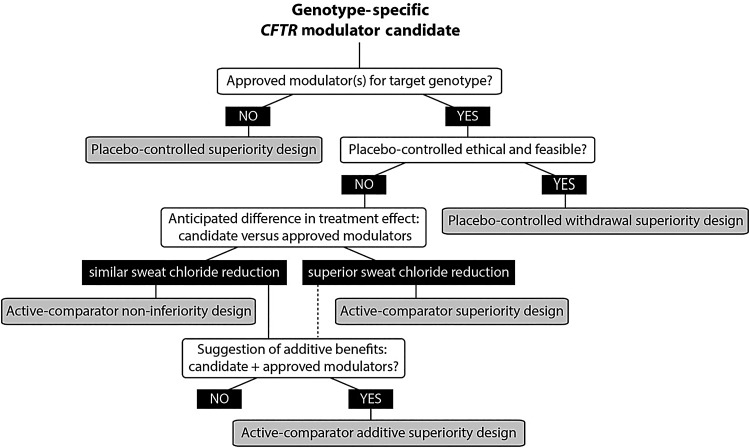
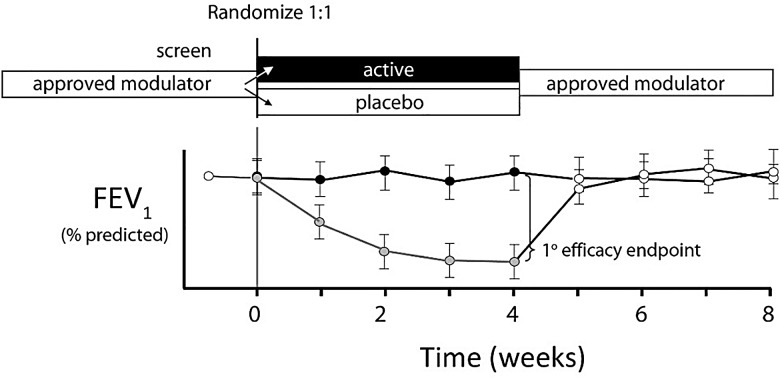
Cystic fibrosis (CF) is a life-shortening genetic disease affecting approximately 70,000 individuals worldwide. Until recently, drug development efforts have emphasised therapies treating downstream signs and symptoms resulting from the underlying CF biological defect: reduced function of the CF transmembrane conductance regulator (CFTR) protein. The current CF drug development landscape has expanded to include therapies that enhance CFTR function by either restoring wild-type CFTR protein expression or increasing (modulating) the function of mutant CFTR proteins in cells. To date, two systemic small-molecule CFTR modulators have been evaluated in pivotal clinical trials in individuals with CF and specific mutant CFTR genotypes that have led to regulatory review and/or approval. Advances in the discovery of CFTR modulators as a promising new class of therapies have been impressive, yet work remains to develop highly effective, disease-modifying modulators for individuals of all CF genotypes. The objectives of this review are to outline the challenges and opportunities in drug development created by systemic genotype-specific CFTR modulators, highlight the advantages of sweat chloride as an established biomarker of CFTR activity to streamline early-phase development and summarise options for later phase clinical trial designs that respond to the adoption of approved genotype-specific modulators into standard of care. An optimal development framework will be needed to move the most promising therapies efficiently through the drug development pipeline and ultimately deliver efficacious and safe therapies to all individuals with CF.
Keywords: Cystic Fibrosis; Rare lung diseases.
Published by the BMJ Publishing Group Limited. For permission to use (where not already granted under a licence) please go to http://www.bmj.com/company/products-services/rights-and-licensing/
Figures
References
-
- Ramsey BW, Pepe MS, Quan JM, et al. . Intermittent administration of inhaled tobramycin in patients with cystic fibrosis. Cystic Fibrosis Inhaled Tobramycin Study Group. N Engl J Med 1999;340:23–30. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical