

Endothelial Plasticity: Shifting Phenotypes through Force Feedback
- PMID: 26904133
- PMCID: PMC4745942
- DOI: 10.1155/2016/9762959
Endothelial Plasticity: Shifting Phenotypes through Force Feedback
Abstract
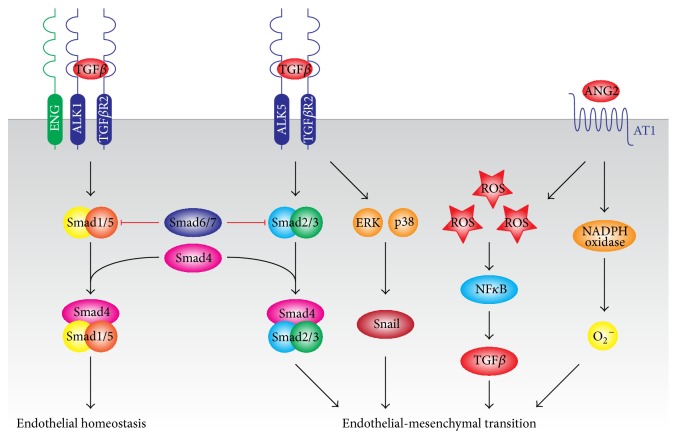
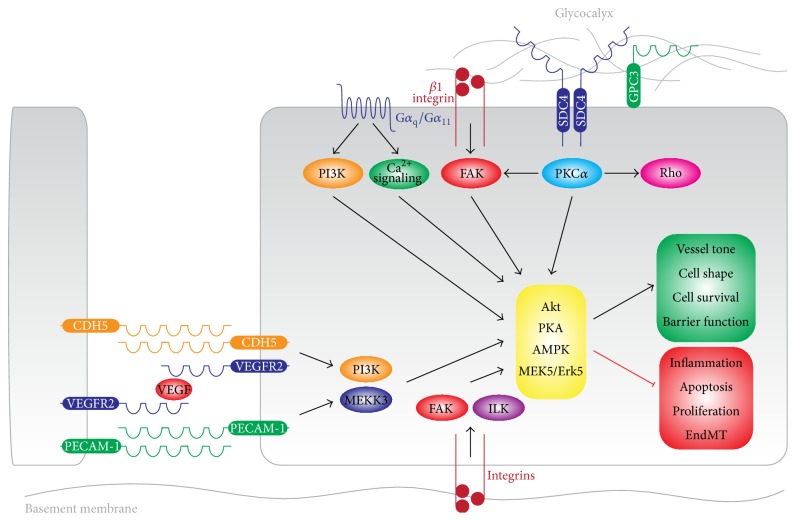
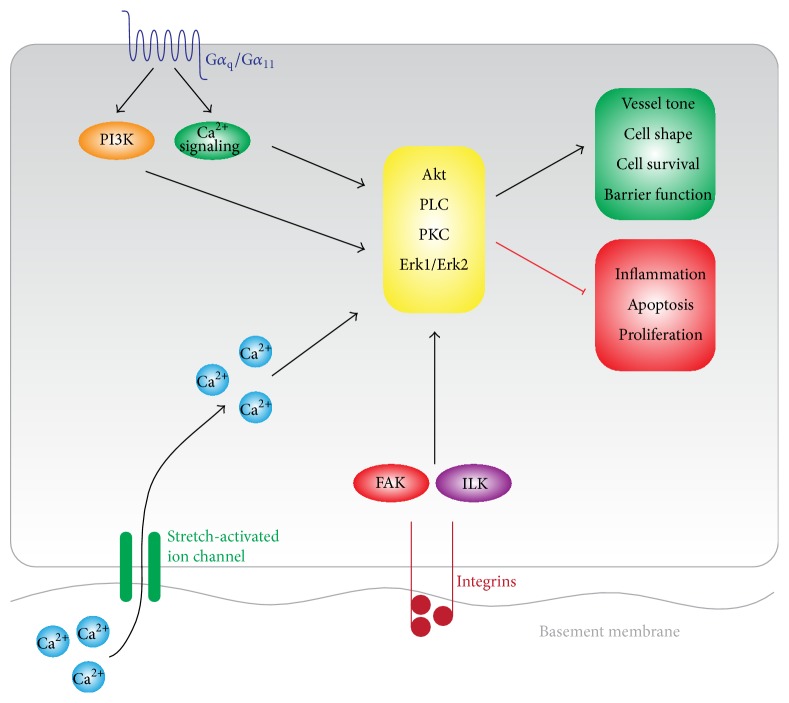
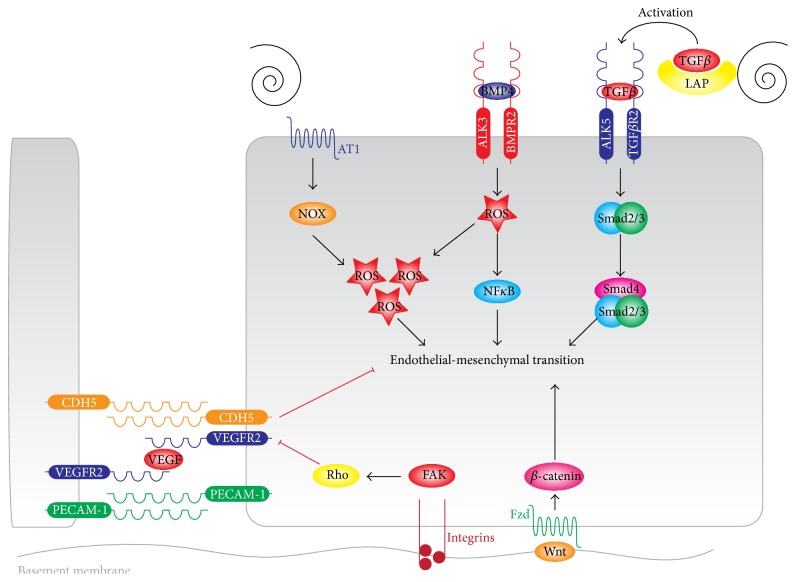
The endothelial lining of the vasculature is exposed to a large variety of biochemical and hemodynamic stimuli with different gradients throughout the vascular network. Adequate adaptation requires endothelial cells to be highly plastic, which is reflected by the remarkable heterogeneity of endothelial cells in tissues and organs. Hemodynamic forces such as fluid shear stress and cyclic strain are strong modulators of the endothelial phenotype and function. Although endothelial plasticity is essential during development and adult physiology, proatherogenic stimuli can induce adverse plasticity which contributes to disease. Endothelial-to-mesenchymal transition (EndMT), the hallmark of endothelial plasticity, was long thought to be restricted to embryonic development but has emerged as a pathologic process in a plethora of diseases. In this perspective we argue how shear stress and cyclic strain can modulate EndMT and discuss how this is reflected in atherosclerosis and pulmonary arterial hypertension.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
