

FAT1 mutations cause a glomerulotubular nephropathy
- PMID: 26905694
- PMCID: PMC4770090
- DOI: 10.1038/ncomms10822
FAT1 mutations cause a glomerulotubular nephropathy
Abstract
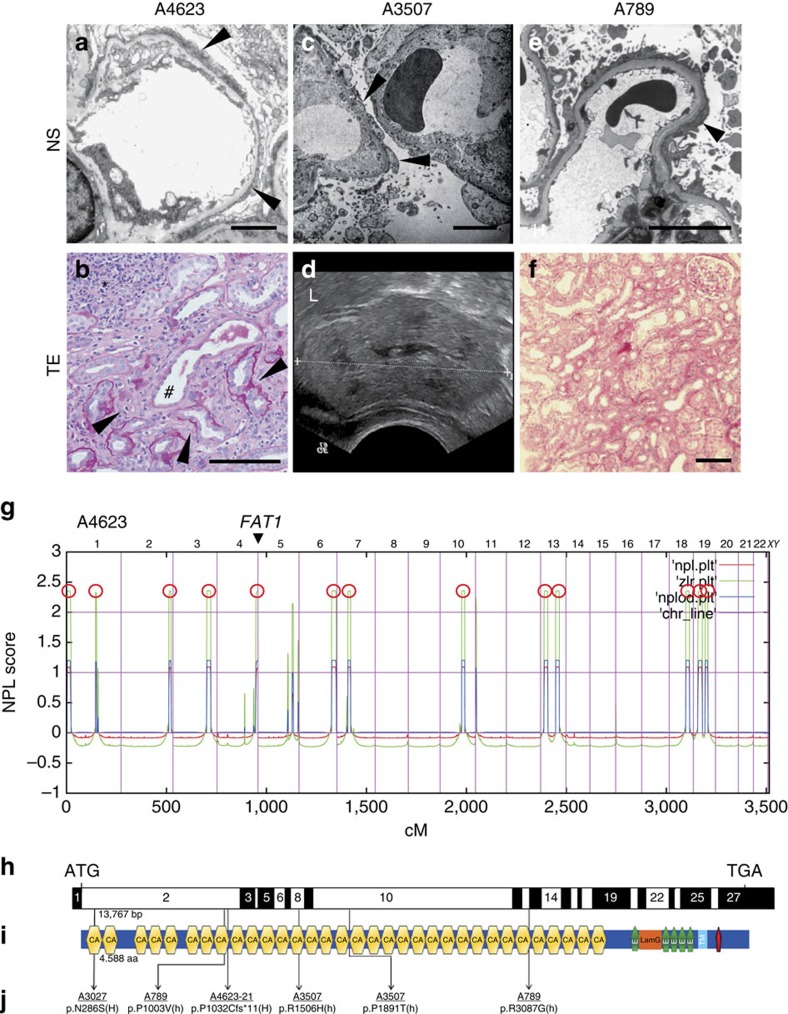
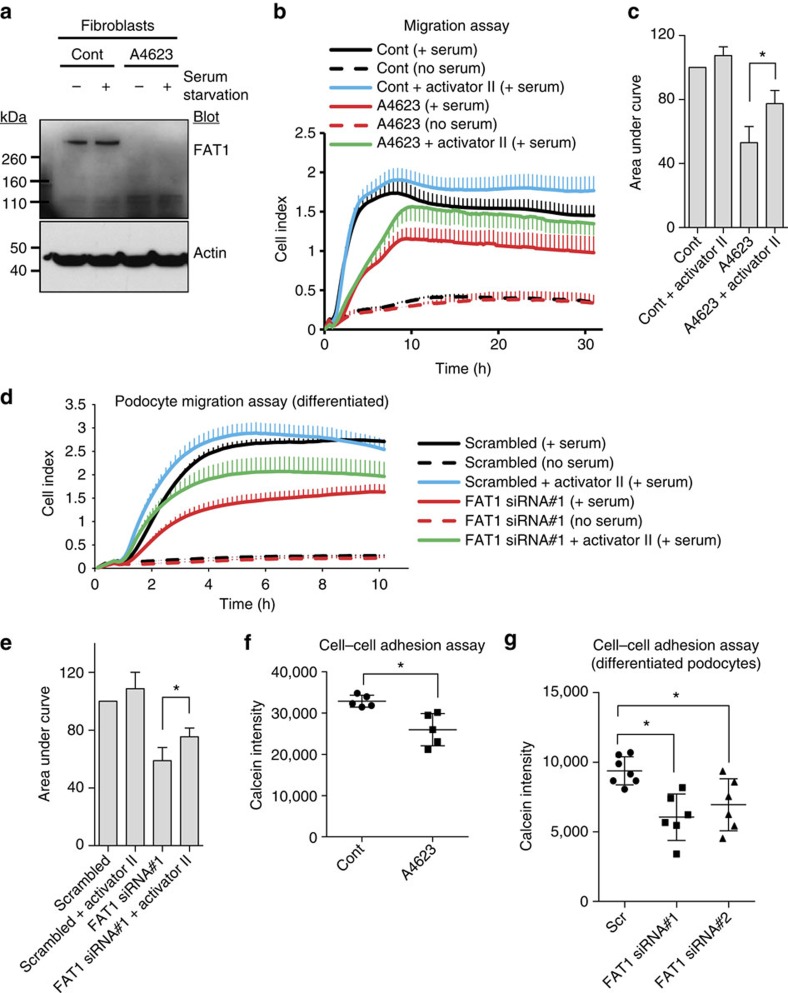
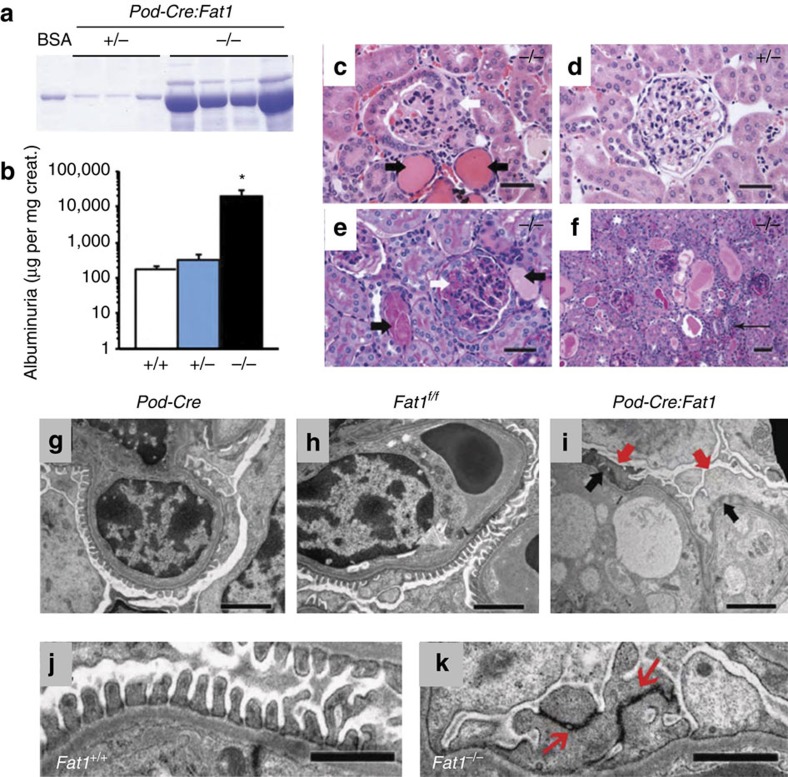
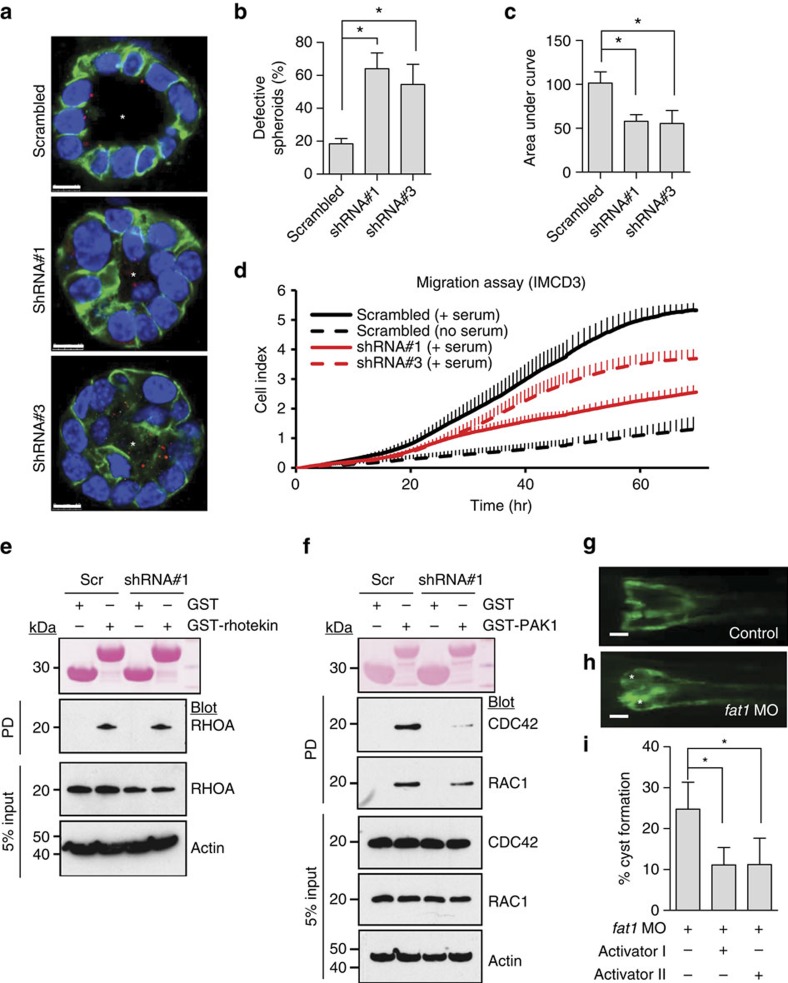
Steroid-resistant nephrotic syndrome (SRNS) causes 15% of chronic kidney disease (CKD). Here we show that recessive mutations in FAT1 cause a distinct renal disease entity in four families with a combination of SRNS, tubular ectasia, haematuria and facultative neurological involvement. Loss of FAT1 results in decreased cell adhesion and migration in fibroblasts and podocytes and the decreased migration is partially reversed by a RAC1/CDC42 activator. Podocyte-specific deletion of Fat1 in mice induces abnormal glomerular filtration barrier development, leading to podocyte foot process effacement. Knockdown of Fat1 in renal tubular cells reduces migration, decreases active RAC1 and CDC42, and induces defects in lumen formation. Knockdown of fat1 in zebrafish causes pronephric cysts, which is partially rescued by RAC1/CDC42 activators, confirming a role of the two small GTPases in the pathogenesis. These findings provide new insights into the pathogenesis of SRNS and tubulopathy, linking FAT1 and RAC1/CDC42 to podocyte and tubular cell function.
Figures
References
-
- Smith J. M., Stablein D. M., Munoz R., Hebert D. & McDonald R. A. Contributions of the transplant registry: The 2006 Annual Report of the North American Pediatric Renal Trials and Collaborative Studies (NAPRTCS). Pediatr. Transplant. 11, 366–373 (2007). - PubMed
-
- Ruf R. G. et al.. Patients with mutations in NPHS2 (podocin) do not respond to standard steroid treatment of nephrotic syndrome. J. Am. Soc. Nephrol. 15, 722–732 (2004). - PubMed
-
- Hildebrandt F. & Heeringa S. F. Specific podocin mutations determine age of onset of nephrotic syndrome all the way into adult life. Kidney Int. 75, 669–671 (2009). - PubMed
-
- Somlo S. & Mundel P. Getting a foothold in nephrotic syndrome. Nat. Genet. 24, 333–335 (2000). - PubMed
-
- Tryggvason K., Patrakka J. & Wartiovaara J. Hereditary proteinuria syndromes and mechanisms of proteinuria. N. Engl. J. Med. 354, 1387–1401 (2006). - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
- G0800571/MRC_/Medical Research Council/United Kingdom
- R01 DK080751/DK/NIDDK NIH HHS/United States
- DK076683/DK/NIDDK NIH HHS/United States
- R01 HL104518/HL/NHLBI NIH HHS/United States
- P30 DK079310/DK/NIDDK NIH HHS/United States
- MR/J006742/1/MRC_/Medical Research Council/United Kingdom
- R01 DK076683/DK/NIDDK NIH HHS/United States
- P30 CA008748/CA/NCI NIH HHS/United States
- R01 DK068306/DK/NIDDK NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- DK090917/DK/NIDDK NIH HHS/United States
- P30 CA013330/CA/NCI NIH HHS/United States
- UM1 HG006504/HG/NHGRI NIH HHS/United States
- DK068306/DK/NIDDK NIH HHS/United States
- K08 DE024774/DE/NIDCR NIH HHS/United States
- HL104518/HL/NHLBI NIH HHS/United States
- RC4 DK090917/DK/NIDDK NIH HHS/United States
- R01 DK059333/DK/NIDDK NIH HHS/United States
- R01 DK098824/DK/NIDDK NIH HHS/United States
- DK059333/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
