

Impaired Lysosomal Integral Membrane Protein 2-dependent Peroxiredoxin 6 Delivery to Lamellar Bodies Accounts for Altered Alveolar Phospholipid Content in Adaptor Protein-3-deficient pearl Mice
- PMID: 26907692
- PMCID: PMC4861416
- DOI: 10.1074/jbc.M116.720201
Impaired Lysosomal Integral Membrane Protein 2-dependent Peroxiredoxin 6 Delivery to Lamellar Bodies Accounts for Altered Alveolar Phospholipid Content in Adaptor Protein-3-deficient pearl Mice
Abstract
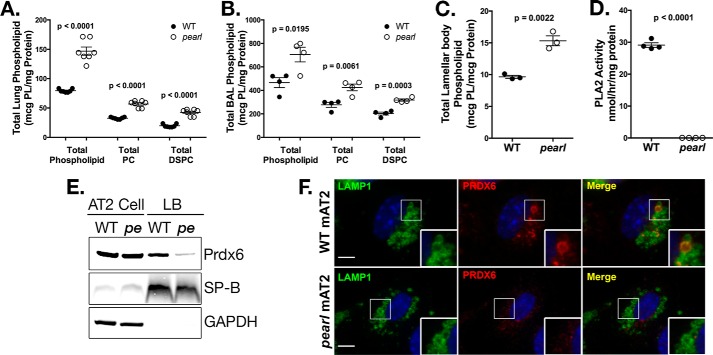
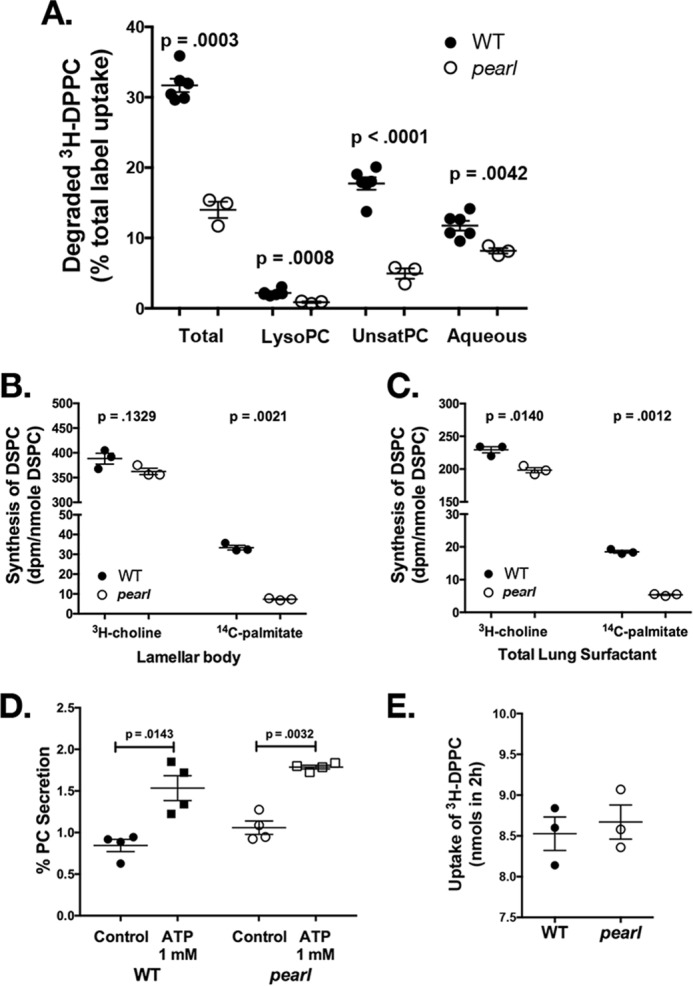
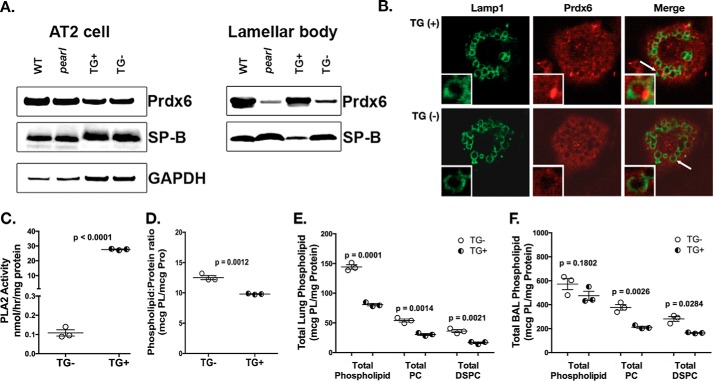
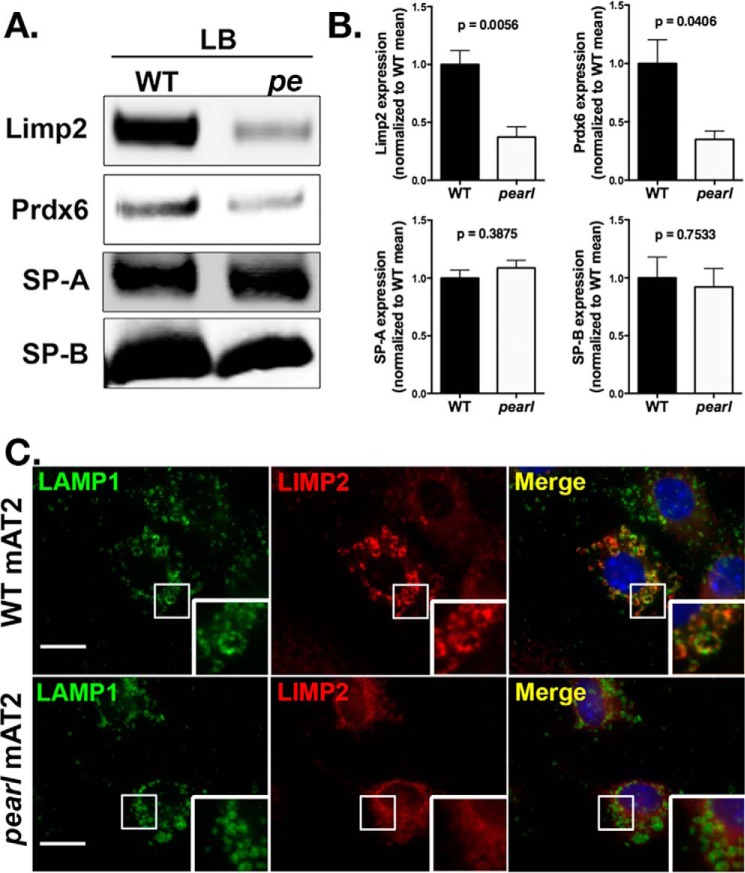
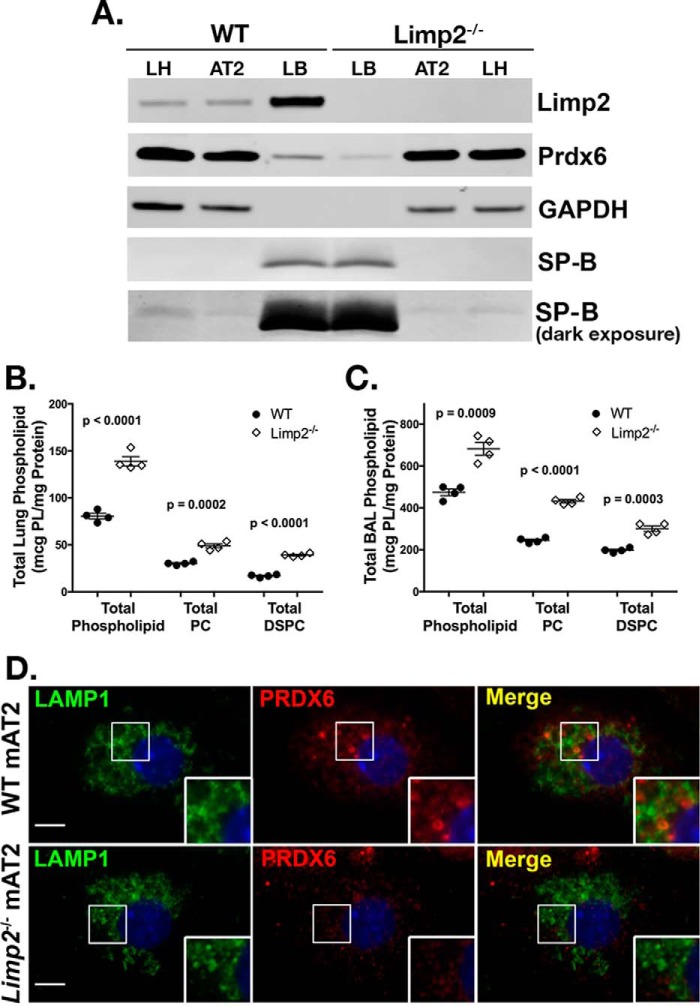
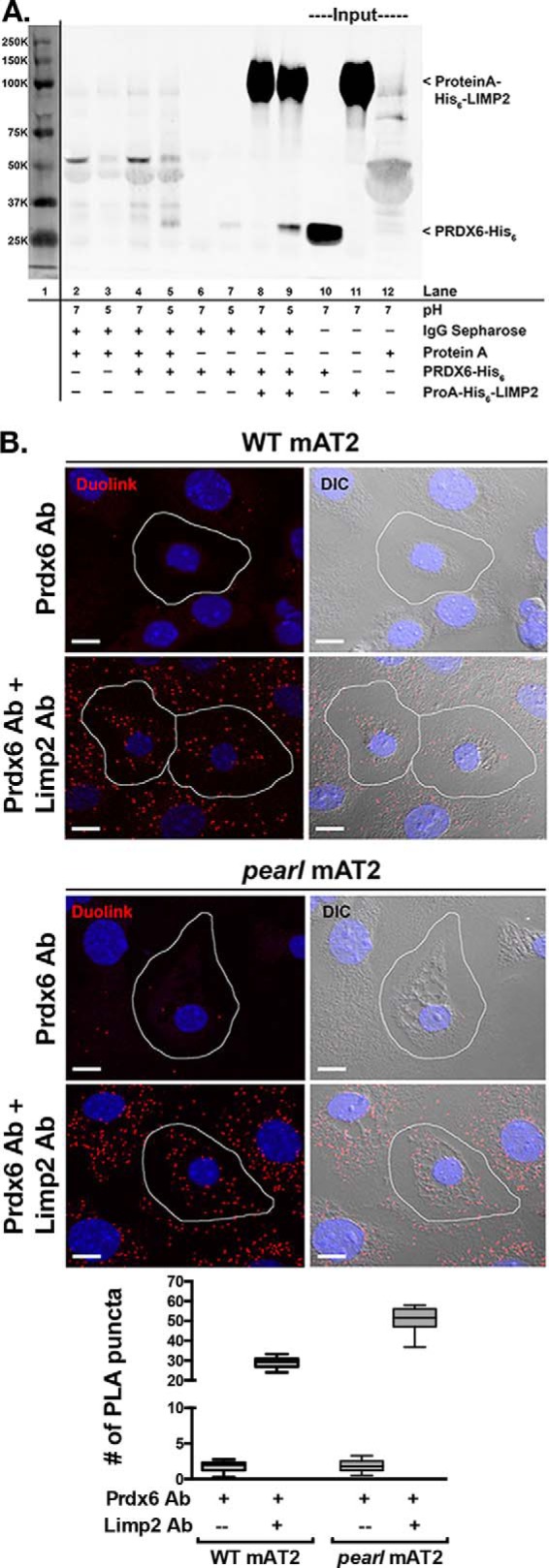
The Hermansky Pudlak syndromes (HPS) constitute a family of disorders characterized by oculocutaneous albinism and bleeding diathesis, often associated with lethal lung fibrosis. HPS results from mutations in genes of membrane trafficking complexes that facilitate delivery of cargo to lysosome-related organelles. Among the affected lysosome-related organelles are lamellar bodies (LB) within alveolar type 2 cells (AT2) in which surfactant components are assembled, modified, and stored. AT2 from HPS patients and mouse models of HPS exhibit enlarged LB with increased phospholipid content, but the mechanism underlying these defects is unknown. We now show that AT2 in the pearl mouse model of HPS type 2 lacking the adaptor protein 3 complex (AP-3) fails to accumulate the soluble enzyme peroxiredoxin 6 (PRDX6) in LB. This defect reflects impaired AP-3-dependent trafficking of PRDX6 to LB, because pearl mouse AT2 cells harbor a normal total PRDX6 content. AP-3-dependent targeting of PRDX6 to LB requires the transmembrane protein LIMP-2/SCARB2, a known AP-3-dependent cargo protein that functions as a carrier for lysosomal proteins in other cell types. Depletion of LB PRDX6 in AP-3- or LIMP-2/SCARB2-deficient mice correlates with phospholipid accumulation in lamellar bodies and with defective intraluminal degradation of LB disaturated phosphatidylcholine. Furthermore, AP-3-dependent LB targeting is facilitated by protein/protein interaction between LIMP-2/SCARB2 and PRDX6 in vitro and in vivo Our data provide the first evidence for an AP-3-dependent cargo protein required for the maturation of LB in AT2 and suggest that the loss of PRDX6 activity contributes to the pathogenic changes in LB phospholipid homeostasis found HPS2 patients.
Keywords: AP-3; Hermansky Pudlak syndrome; adaptor protein; alveolar epithelial type 2 cell; lamellar body; lung; peroxiredoxin; peroxiredoxin 6; phospholipid metabolism; trafficking.
© 2016 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
References
-
- Wei M. L. (2006) Hermansky-Pudlak syndrome: a disease of protein trafficking and organelle function. Pigment Cell Res. 19, 19–42 - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
