

Mutation allele burden remains unchanged in chronic myelomonocytic leukaemia responding to hypomethylating agents
- PMID: 26908133
- PMCID: PMC4770084
- DOI: 10.1038/ncomms10767
Mutation allele burden remains unchanged in chronic myelomonocytic leukaemia responding to hypomethylating agents
Abstract
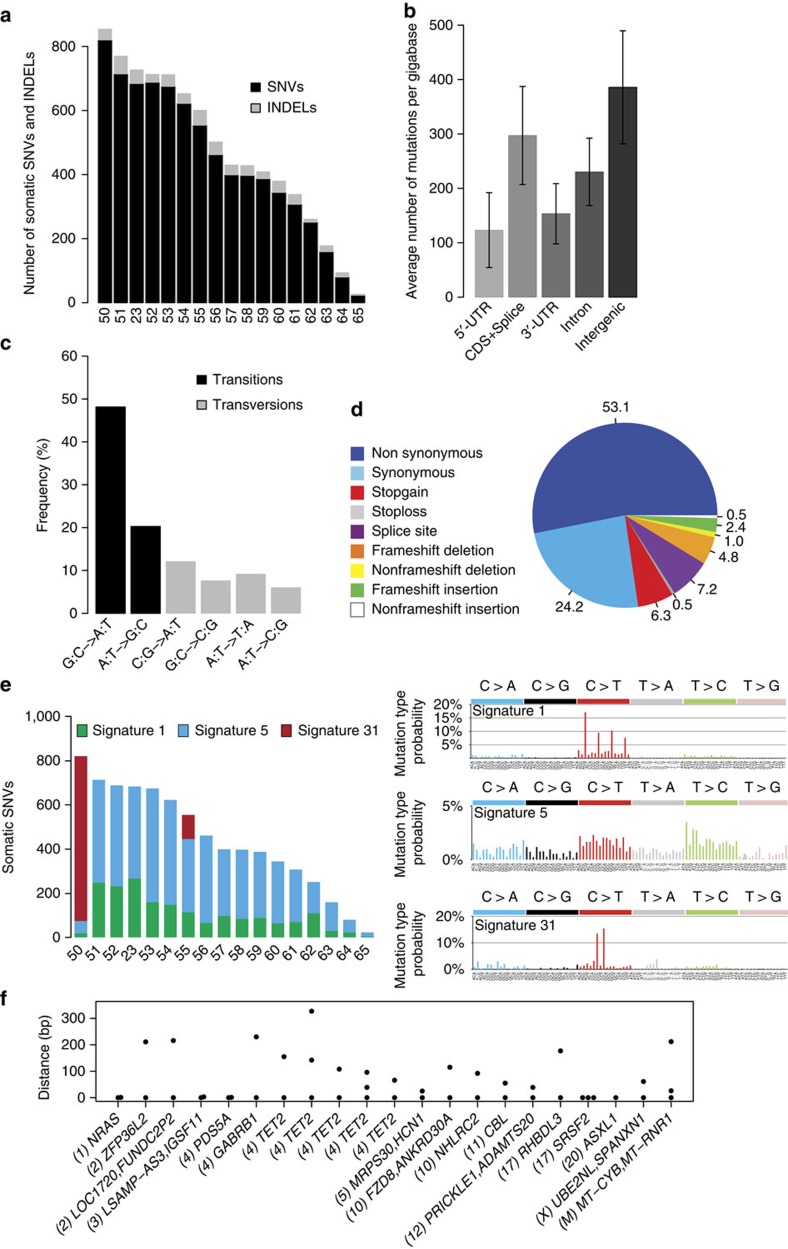
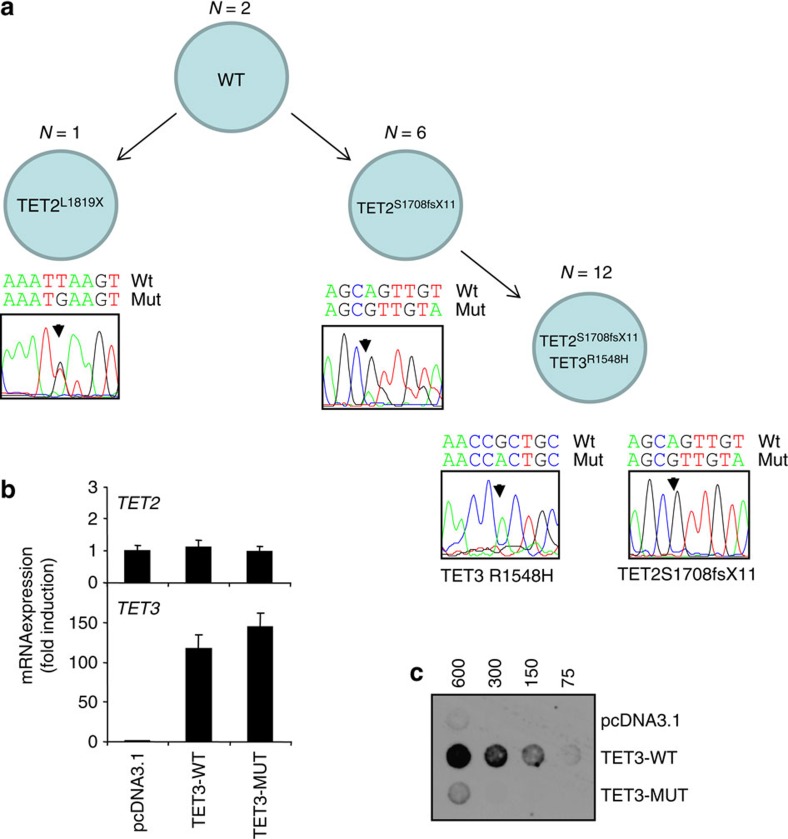
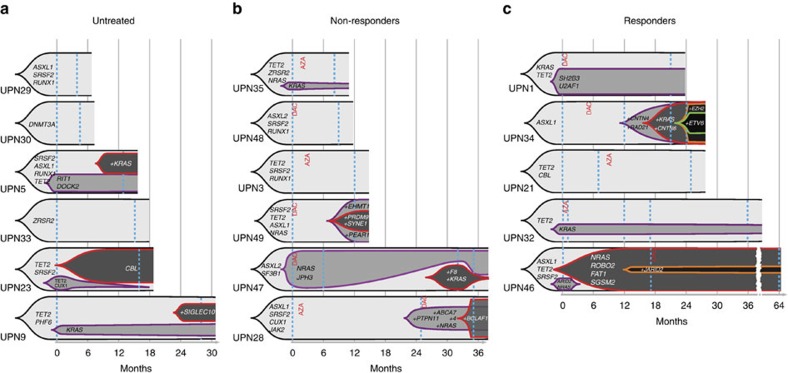
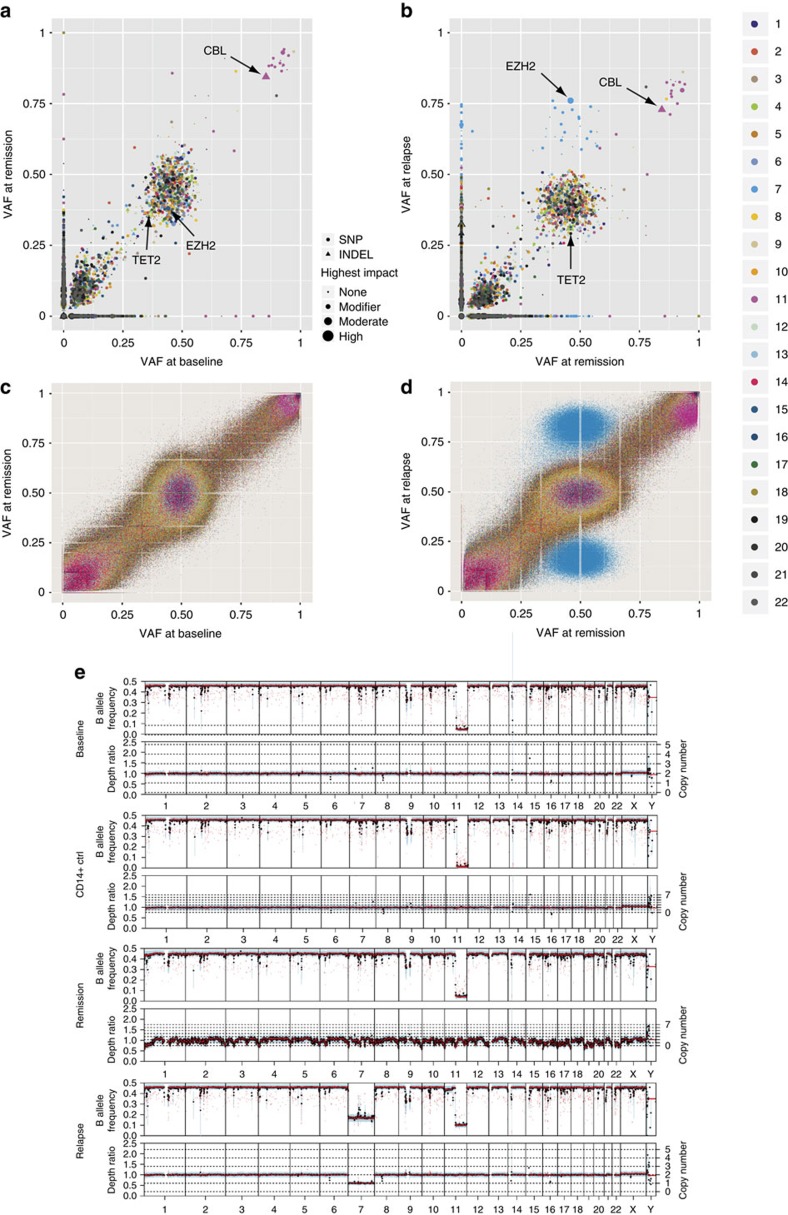
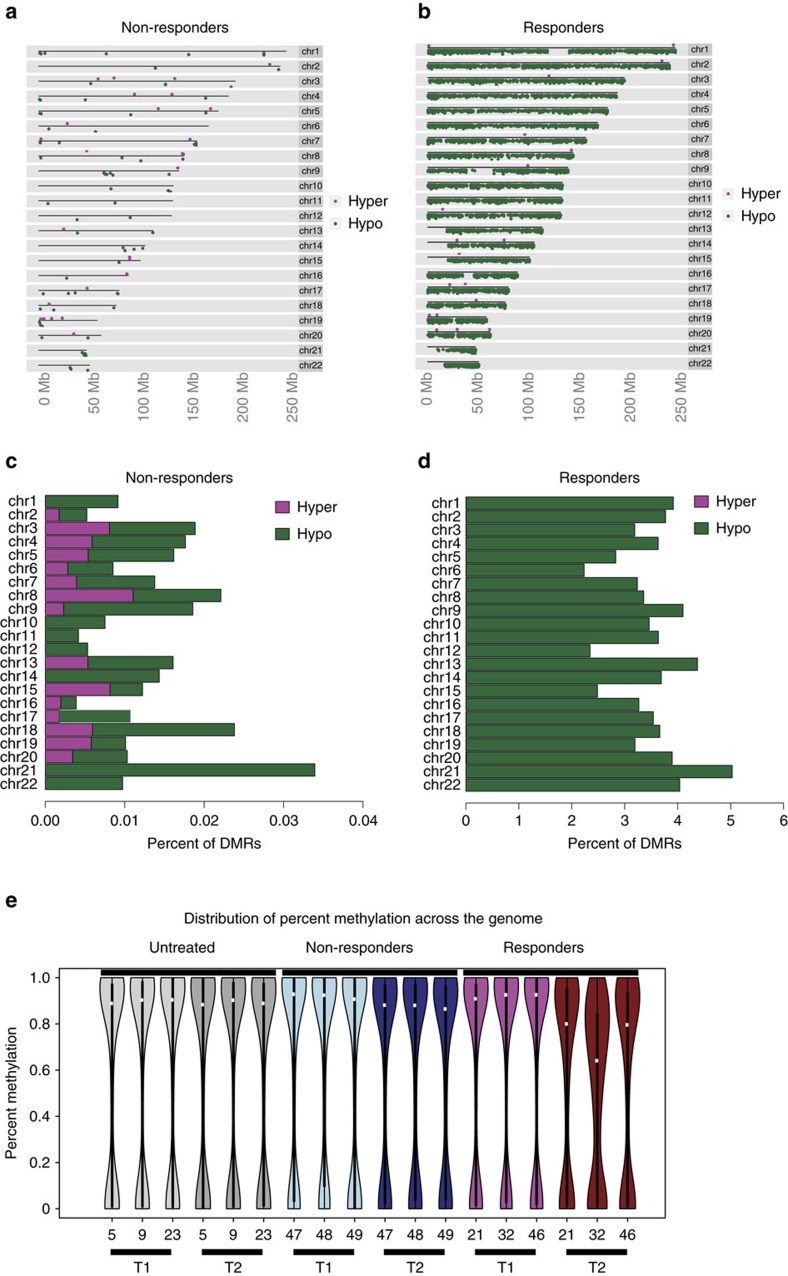
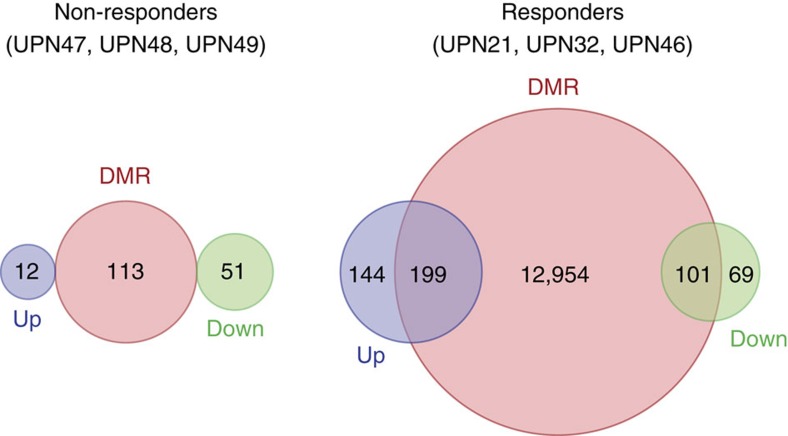
The cytidine analogues azacytidine and 5-aza-2'-deoxycytidine (decitabine) are commonly used to treat myelodysplastic syndromes, with or without a myeloproliferative component. It remains unclear whether the response to these hypomethylating agents results from a cytotoxic or an epigenetic effect. In this study, we address this question in chronic myelomonocytic leukaemia. We describe a comprehensive analysis of the mutational landscape of these tumours, combining whole-exome and whole-genome sequencing. We identify an average of 14±5 somatic mutations in coding sequences of sorted monocyte DNA and the signatures of three mutational processes. Serial sequencing demonstrates that the response to hypomethylating agents is associated with changes in DNA methylation and gene expression, without any decrease in the mutation allele burden, nor prevention of new genetic alteration occurence. Our findings indicate that cytosine analogues restore a balanced haematopoiesis without decreasing the size of the mutated clone, arguing for a predominantly epigenetic effect.
Figures


References
-
- Vardiman J. W. et al.. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood 114, 937–951 (2009). - PubMed
-
- Padron E. & Steensma D. P. Cutting the cord from myelodysplastic syndromes: chronic myelomonocytic leukemia-specific biology and management strategies. Curr. Opin. Hematol. 22, 163–170 (2015). - PubMed
-
- Itzykson R. et al.. Clonal architecture of chronic myelomonocytic leukemias. Blood 121, 2186–2198 (2013). - PubMed
-
- Kon A. et al.. Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat. Genet. 45, 1232–1237 (2013). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
