

Allelic series of Huntington's disease knock-in mice reveals expression discorrelates
- PMID: 26908599
- PMCID: PMC4805312
- DOI: 10.1093/hmg/ddw040
Allelic series of Huntington's disease knock-in mice reveals expression discorrelates
Abstract
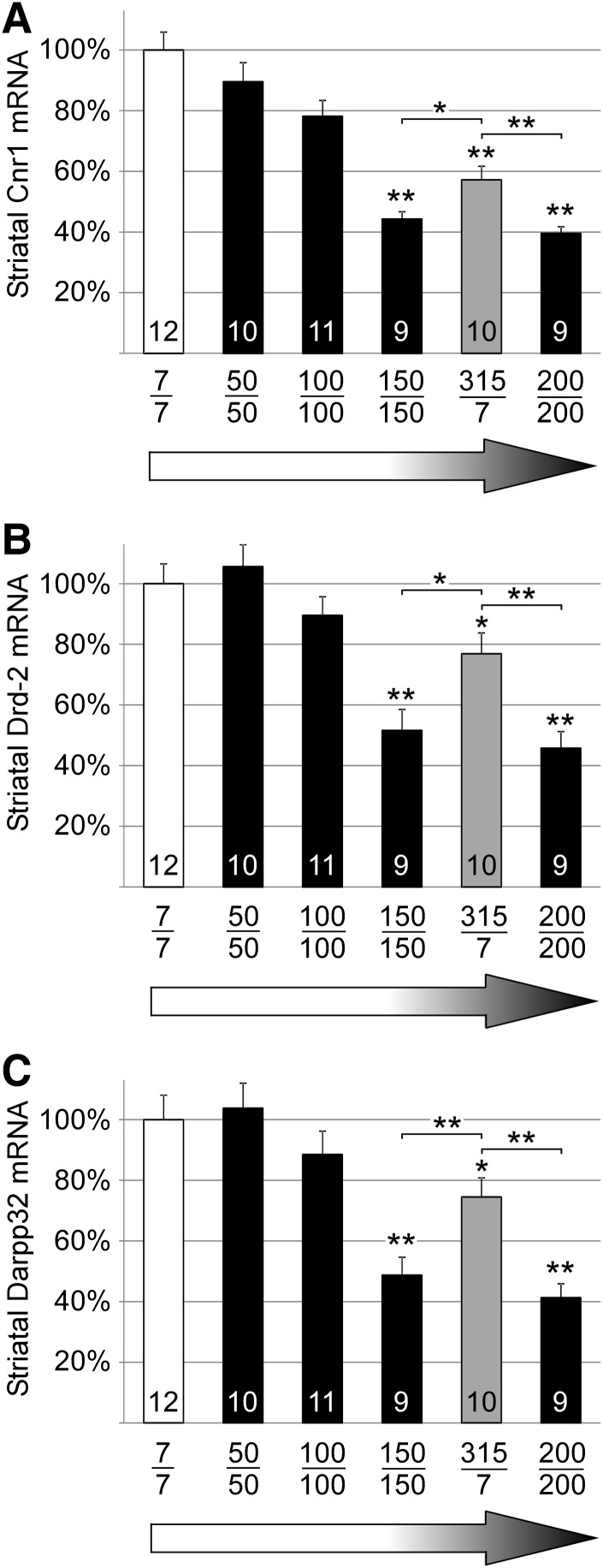
Identifying molecular drivers of pathology provides potential therapeutic targets. Differentiating between drivers and coincidental molecular alterations presents a major challenge. Variation unrelated to pathology further complicates transcriptomic, proteomic and metabolomic studies which measure large numbers of individual molecules. To overcome these challenges towards the goal of determining drivers of Huntington's disease (HD), we generated an allelic series of HD knock-in mice with graded levels of phenotypic severity for comparison with molecular alterations. RNA-sequencing analysis of this series reveals high numbers of transcripts with level alterations that do not correlate with phenotypic severity. These discorrelated molecular changes are unlikely to be drivers of pathology allowing an exclusion-based strategy to provide a short list of driver candidates. Further analysis of the data shows that a majority of transcript level changes in HD knock-in mice involve alteration of the rate of mRNA processing and/or degradation rather than solely being due to alteration of transcription rate. The overall strategy described can be applied to assess the influence of any molecular change on pathology for diseases where different mutations cause graded phenotypic severity.
© The Author 2016. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures






References
-
- Huntington's Disease Collaborative Research Group. (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell, 72, 971–983. - PubMed
-
- Cummings D.M., Alaghband Y., Hickey M.A., Joshi P.R., Hong S.C., Zhu C., Ando T.K., Andre V.M., Cepeda C., Watson J.B. et al. (2012) A critical window of CAG repeat-length correlates with phenotype severity in the R6/2 mouse model of Huntington's disease. J. Neurophysiol., 107, 677–691. - PMC - PubMed
-
- Morton A.J., Glynn D., Leavens W., Zheng Z., Faull R.L., Skepper J.N., Wight J.M. (2009) Paradoxical delay in the onset of disease caused by super-long CAG repeat expansions in R6/2 mice. Neurobiol. Dis., 33, 331–341. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
