

The role of decidual cells in uterine hemostasis, menstruation, inflammation, adverse pregnancy outcomes and abnormal uterine bleeding
- PMID: 26912000
- PMCID: PMC4917742
- DOI: 10.1093/humupd/dmw004
The role of decidual cells in uterine hemostasis, menstruation, inflammation, adverse pregnancy outcomes and abnormal uterine bleeding
Abstract
Background: Human pregnancy requires robust hemostasis to prevent hemorrhage during extravillous trophoblast (EVT) invasion of the decidualized endometrium, modification of spiral arteries and post-partum processes. However, decidual hemorrhage (abruption) can occur throughout pregnancy from poorly transformed spiral arteries, causing fetal death or spontaneous preterm birth (PTB), or it can promote the aberrant placentation observed in intrauterine growth restriction (IUGR) and pre-eclampsia; all leading causes of perinatal or maternal morbidity and mortality. In non-fertile cycles, the decidua undergoes controlled menstrual bleeding. Abnormal uterine bleeding (AUB) accompanying progestin-only, long-acting, reversible contraception (pLARC) accounts for most discontinuations of these safe and highly effective agents, thereby contributing to unwanted pregnancies and abortion. The aim of this study was to investigate the role of decidual cells in uterine hemostasis, menstruation, inflammation, adverse pregnancy outcomes and abnormal uterine bleeding.
Methods: We conducted a critical review of the literature arising from PubMed searches up to December 2015, regarding in situ and in vitro expression and regulation of several specific proteins involved in uterine hemostasis in decidua and cycling endometrium. In addition, we discussed clinical and molecular mechanisms associated with pLARC-induced AUB and pregnancy complications with abruptions, chorioamnionitis or pre-eclampsia.
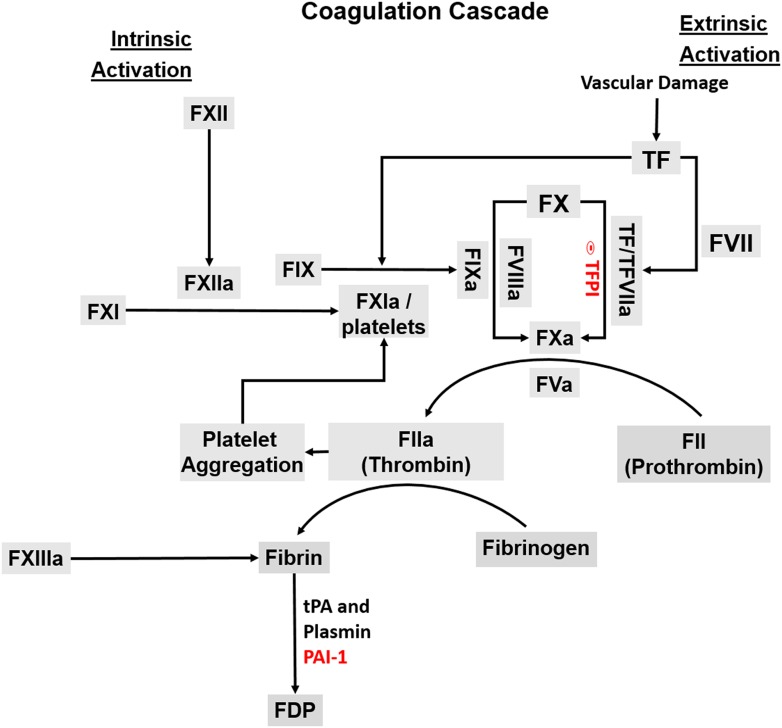
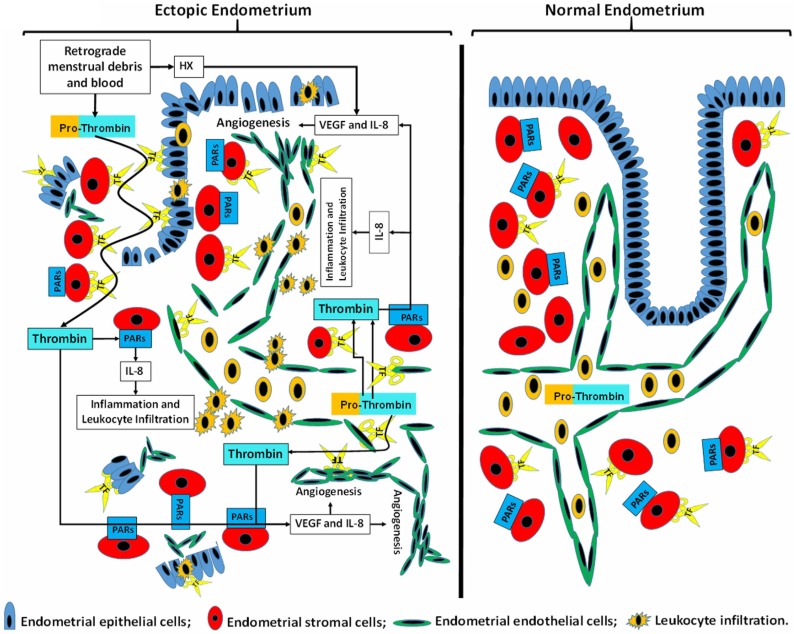
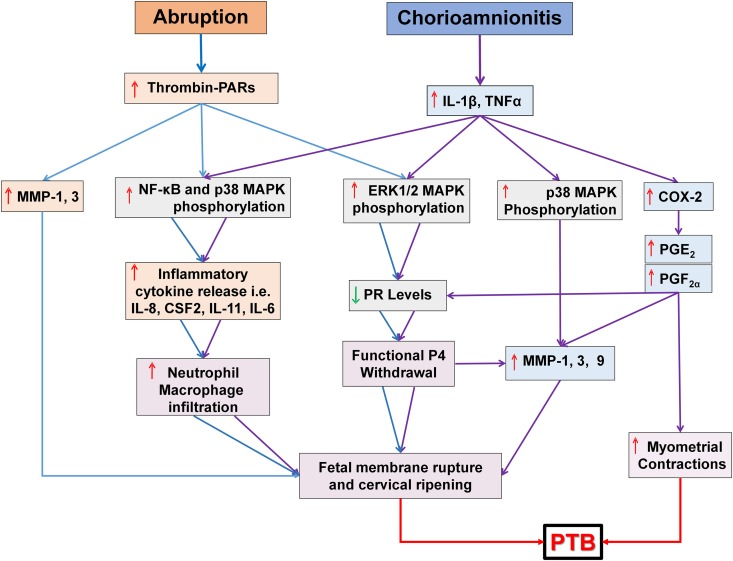
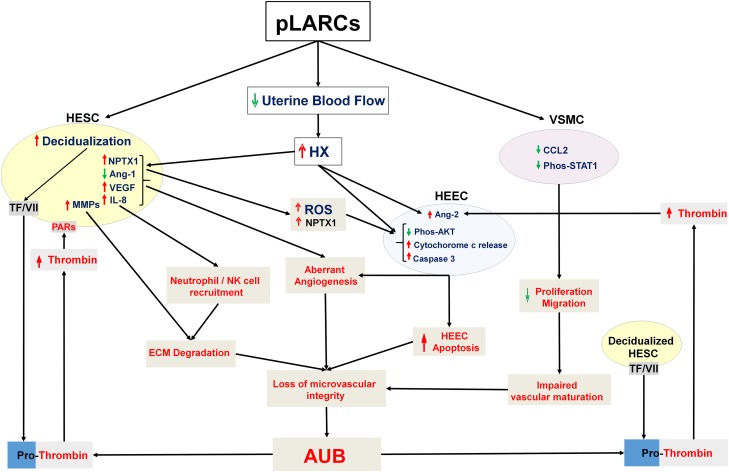
Results: Progestin-induced decidualization of estradiol-primed human endometrial stromal cells (HESCs) increases in vivo and in vitro expression of tissue factor (TF) and type-1 plasminogen activator inhibitor (PAI-1) while inhibiting plasminogen activators (PAs), matrix metalloproteinases (MMPs), and the vasoconstrictor, endothelin-1 (ET-1). These changes in decidual cell-derived regulators of hemostasis, fibrinolysis, extracellular matrix (ECM) turnover, and vascular tone prevent hemorrhage during EVT invasion and vascular remodeling. In non-fertile cycles, progesterone withdrawal reduces TF and PAI-1 while increasing PA, MMPs and ET-1, causing menstrual-associated bleeding, fibrinolysis, ECM degradation and ischemia. First trimester decidual hemorrhage elicits later adverse outcomes including pregnancy loss, pre-eclampsia, abruption, IUGR and PTB. Decidual hemorrhage generates excess thrombin that binds to decidual cell-expressed protease-activated receptors (PARs) to induce chemokines promoting shallow placentation; such bleeding later in pregnancy generates thrombin to down-regulate decidual cell progesterone receptors and up-regulate cytokines and MMPs linked to PTB. Endometria of pLARC users display ischemia-induced excess vasculogenesis and progestin inhibition of spiral artery vascular smooth muscle cell proliferation and migration leading to dilated fragile vessels prone to bleeding. Moreover, aberrant TF-derived thrombin signaling also contributes to the pathogenesis of endometriosis via induction of angiogenesis, inflammation and cell survival.
Conclusion: Perivascular decidualized HESCs promote endometrial hemostasis during placentation yet facilitate menstruation through progestational regulation of hemostatic, proteolytic, and vasoactive proteins. Pathological endometrial hemorrhage elicits excess local thrombin generation, which contributes to pLARC associated AUB, endometriosis and adverse pregnancy outcomes through several biochemical mechanisms.
Keywords: contraception; decidual cells; pre-eclampsia; preterm birth; thrombin; tissue factor; uterine bleeding.
© The Author 2016. Published by Oxford University Press on behalf of the European Society of Human Reproduction and Embryology. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures
References
-
- Affandi B. An integrated analysis of vaginal bleeding patterns in clinical trials of Implanon. Contraception 1998;58:99S–107S. - PubMed
-
- Ahamed J, Ruf W. Protease-activated receptor 2-dependent phosphorylation of the tissue factor cytoplasmic domain. J Biol Chem 2004;279:23038–23044. - PubMed
-
- Albrecht ED, Pepe GJ. Steroid hormone regulation of angiogenesis in the primate endometrium. Front Biosci 2003;8:d416–d429. - PubMed
-
- Aluvihare VR, Kallikourdis M, Betz AG. Regulatory T cells mediate maternal tolerance to the fetus. Nat Immunol 2004;5:266–271. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
