

APOBEC3G and APOBEC3F Act in Concert To Extinguish HIV-1 Replication
- PMID: 26912618
- PMCID: PMC4836340
- DOI: 10.1128/JVI.03275-15
APOBEC3G and APOBEC3F Act in Concert To Extinguish HIV-1 Replication
Abstract
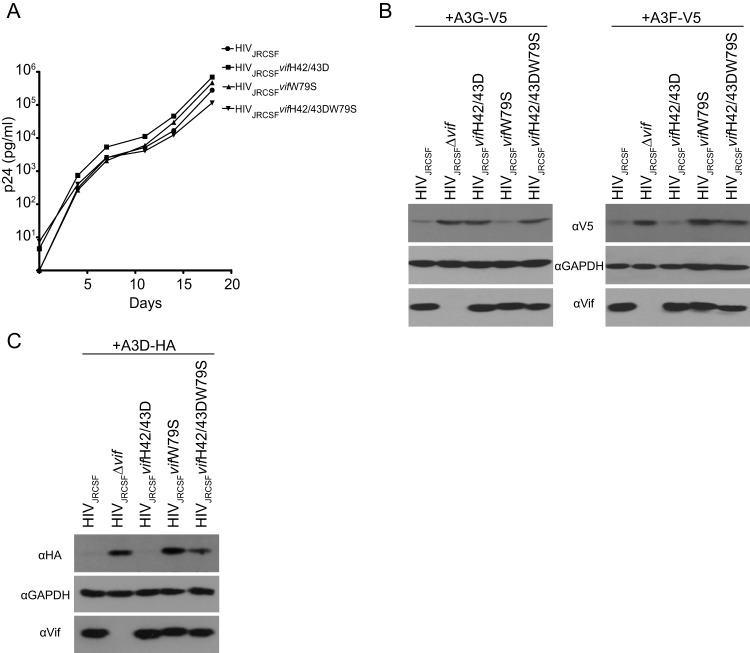
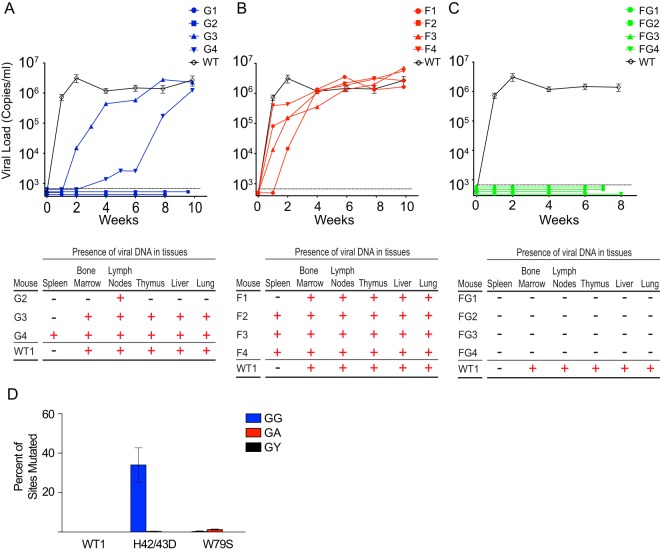
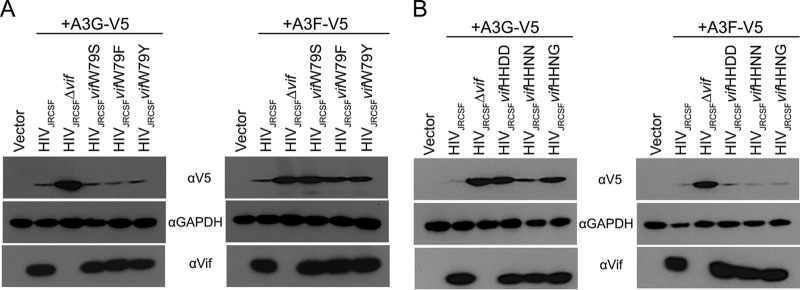
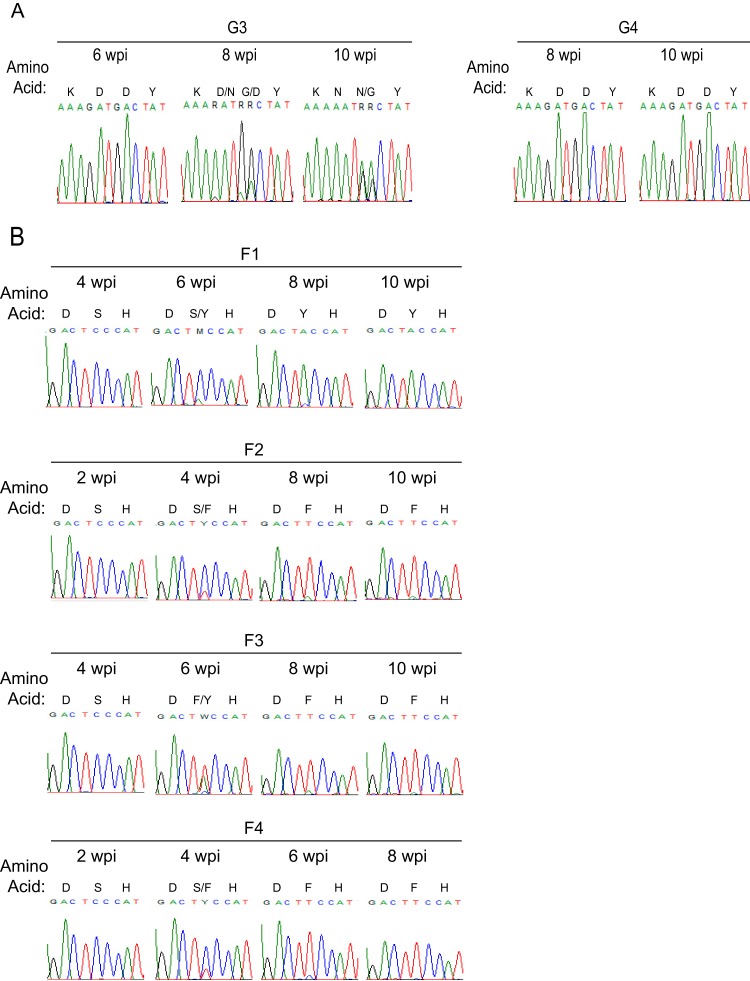
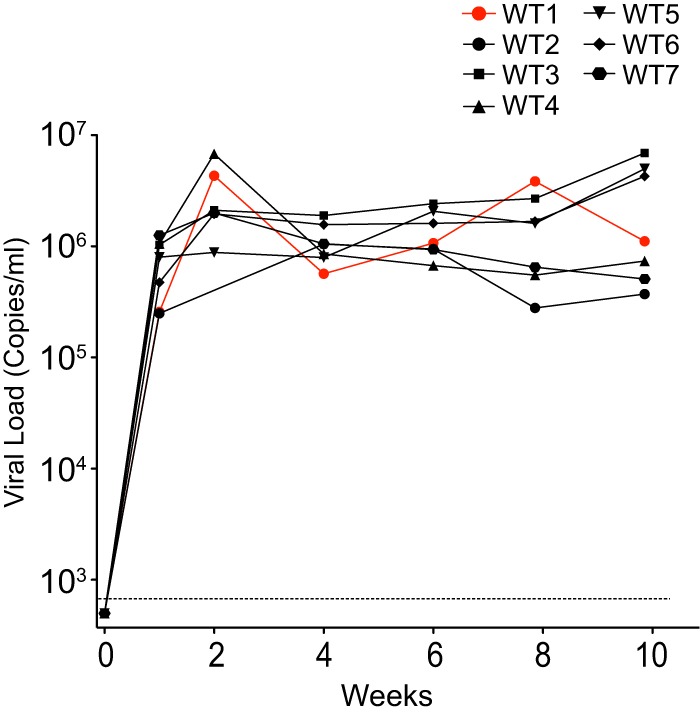
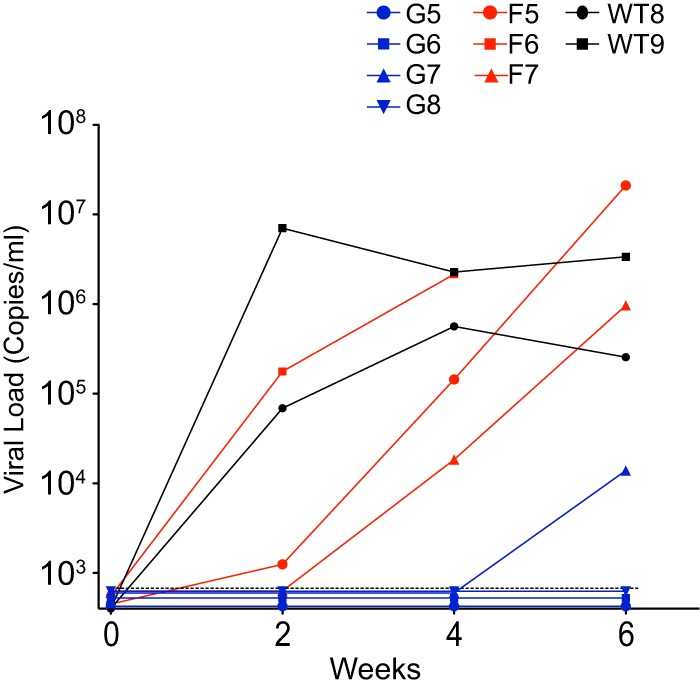
The multifunctional HIV-1 accessory protein Vif counters the antiviral activities of APOBEC3G (A3G) and APOBEC3F (A3F), and some Vifs counter stable alleles of APOBEC3H (A3H). Studies in humanized mice have shown that HIV-1 lacking Vif expression is not viable. Here, we look at the relative contributions of the three APOBEC3s to viral extinction. Inoculation of bone marrow/liver/thymus (BLT) mice with CCR5-tropic HIV-1JRCSF(JRCSF) expressing a vif gene inactive for A3G but not A3F degradation activity (JRCSFvifH42/43D) displayed either no or delayed replication. JRCSF expressing a vif gene mutated to inactivate A3F degradation but not A3G degradation (JRCSFvifW79S) always replicated to high viral loads with variable delays. JRCSF with vif mutated to lack both A3G and A3F degradation activities (JRCSFvifH42/43DW79S) failed to replicate, mimicking JRCSF without Vif expression (JRCSFΔvif). JRCSF and JRCSFvifH42/43D, but not JRCSFvifW79S or JRCSFvifH42/43DW79S, degraded APOBEC3D. With one exception, JRCSFs expressing mutant Vifs that replicated acquired enforced vif mutations. These mutations partially restored A3G or A3F degradation activity and fully replaced JRCSFvifH42/43D or JRCSFvifW79S by 10 weeks. Surprisingly, induced mutations temporally lagged behind high levels of virus in blood. In the exceptional case, JRCSFvifH42/43D replicated after a prolonged delay with no mutations in vif but instead a V27I mutation in the RNase H coding sequence. JRCSFvifH42/43D infections exhibited massive GG/AG mutations in pol viral DNA, but in viral RNA, there were no fixed mutations in the Gag or reverse transcriptase coding sequence. A3H did not contribute to viral extinction but, in combination with A3F, could delay JRCSF replication. A3H was also found to hypermutate viral DNA.
Importance: Vif degradation of A3G and A3F enhances viral fitness, as virus with even a partially restored capacity for degradation outgrows JRCSFvifH42/43D and JRCSFvifW79S. Unexpectedly, fixation of mutations that replaced H42/43D or W79S in viral RNA lagged behind the appearance of high viral loads. In one exceptional JRCSFvifH42/43D infection, vif was unchanged but replication proceeded after a long delay. These results suggest that Vif binds and inhibits the non-cytosine deaminase activities of intact A3G and intact A3F, allowing JRCSFvifH42/43D and JRCSFvifW79S to replicate with reduced fitness. Subsequently, enhanced Vif function is acquired by enforced mutations. In infected cells, JRCSFΔvif and JRCSFvifH42/43DW79S are exposed to active A3F and A3G and fail to replicate. JRCSFvifH42/43D Vif degrades A3F and, in some cases, overcomes A3G mutagenic activity to replicate. Vif may have evolved to inhibit A3F and A3G by stoichiometric binding and subsequently acquired the ability to target these proteins to proteasomes.
Copyright © 2016, American Society for Microbiology. All Rights Reserved.
Figures



Similar articles
-
Mechanism of Enhanced HIV Restriction by Virion Coencapsidated Cytidine Deaminases APOBEC3F and APOBEC3G.J Virol. 2017 Jan 18;91(3):e02230-16. doi: 10.1128/JVI.02230-16. Print 2017 Feb 1. J Virol. 2017. PMID: 27881650 Free PMC article.
-
The activity spectrum of Vif from multiple HIV-1 subtypes against APOBEC3G, APOBEC3F, and APOBEC3H.J Virol. 2012 Jan;86(1):49-59. doi: 10.1128/JVI.06082-11. Epub 2011 Oct 19. J Virol. 2012. PMID: 22013041 Free PMC article.
-
Identification of a critical T(Q/D/E)x5ADx2(I/L) motif from primate lentivirus Vif proteins that regulate APOBEC3G and APOBEC3F neutralizing activity.J Virol. 2010 Sep;84(17):8561-70. doi: 10.1128/JVI.00960-10. Epub 2010 Jun 30. J Virol. 2010. PMID: 20592083 Free PMC article.
-
Evasion of adaptive immunity by HIV through the action of host APOBEC3G/F enzymes.AIDS Res Ther. 2017 Sep 12;14(1):44. doi: 10.1186/s12981-017-0173-8. AIDS Res Ther. 2017. PMID: 28893290 Free PMC article. Review.
-
Various strategies for developing APOBEC3G protectors to circumvent human immunodeficiency virus type 1.Eur J Med Chem. 2023 Mar 15;250:115188. doi: 10.1016/j.ejmech.2023.115188. Epub 2023 Feb 6. Eur J Med Chem. 2023. PMID: 36773550 Review.
Cited by
-
Human Primary Macrophages Derived In Vitro from Circulating Monocytes Comprise Adherent and Non-Adherent Subsets with Differential Expression of Siglec-1 and CD4 and Permissiveness to HIV-1 Infection.Front Immunol. 2017 Oct 23;8:1352. doi: 10.3389/fimmu.2017.01352. eCollection 2017. Front Immunol. 2017. PMID: 29123518 Free PMC article.
-
Human APOBEC3 Variations and Viral Infection.Viruses. 2021 Jul 14;13(7):1366. doi: 10.3390/v13071366. Viruses. 2021. PMID: 34372572 Free PMC article. Review.
-
Impact of Suboptimal APOBEC3G Neutralization on the Emergence of HIV Drug Resistance in Humanized Mice.J Virol. 2020 Feb 14;94(5):e01543-19. doi: 10.1128/JVI.01543-19. Print 2020 Feb 14. J Virol. 2020. PMID: 31801862 Free PMC article.
-
Variability in HIV-1 transmitted/founder virus susceptibility to combined APOBEC3F and APOBEC3G host restriction.J Virol. 2025 Jan 31;99(1):e0160624. doi: 10.1128/jvi.01606-24. Epub 2024 Dec 23. J Virol. 2025. PMID: 39714157 Free PMC article.
-
HIV-1 competition experiments in humanized mice show that APOBEC3H imposes selective pressure and promotes virus adaptation.PLoS Pathog. 2017 May 5;13(5):e1006348. doi: 10.1371/journal.ppat.1006348. eCollection 2017 May. PLoS Pathog. 2017. PMID: 28475648 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
