

Allosteric Inhibition of Bcr-Abl Kinase by High Affinity Monobody Inhibitors Directed to the Src Homology 2 (SH2)-Kinase Interface
- PMID: 26912659
- PMCID: PMC4861451
- DOI: 10.1074/jbc.M115.707901
Allosteric Inhibition of Bcr-Abl Kinase by High Affinity Monobody Inhibitors Directed to the Src Homology 2 (SH2)-Kinase Interface
Abstract
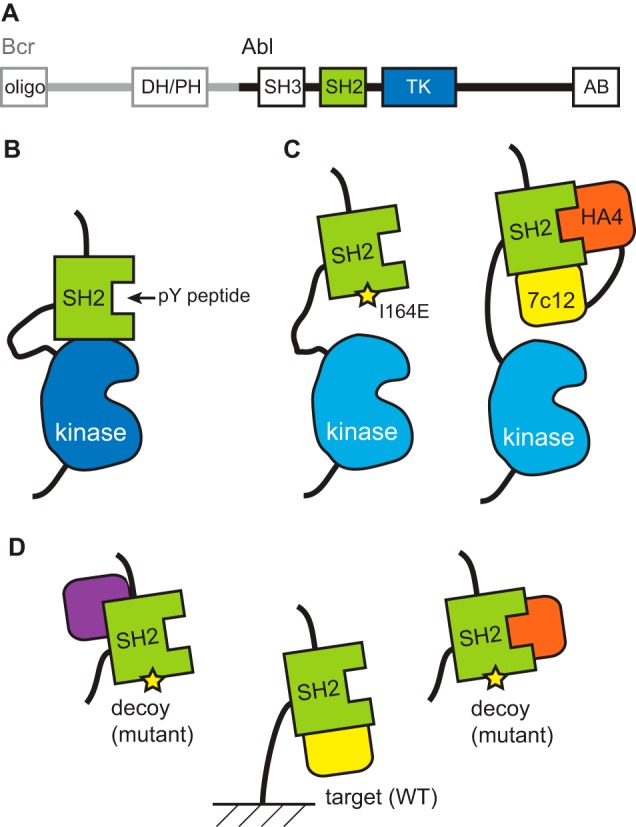
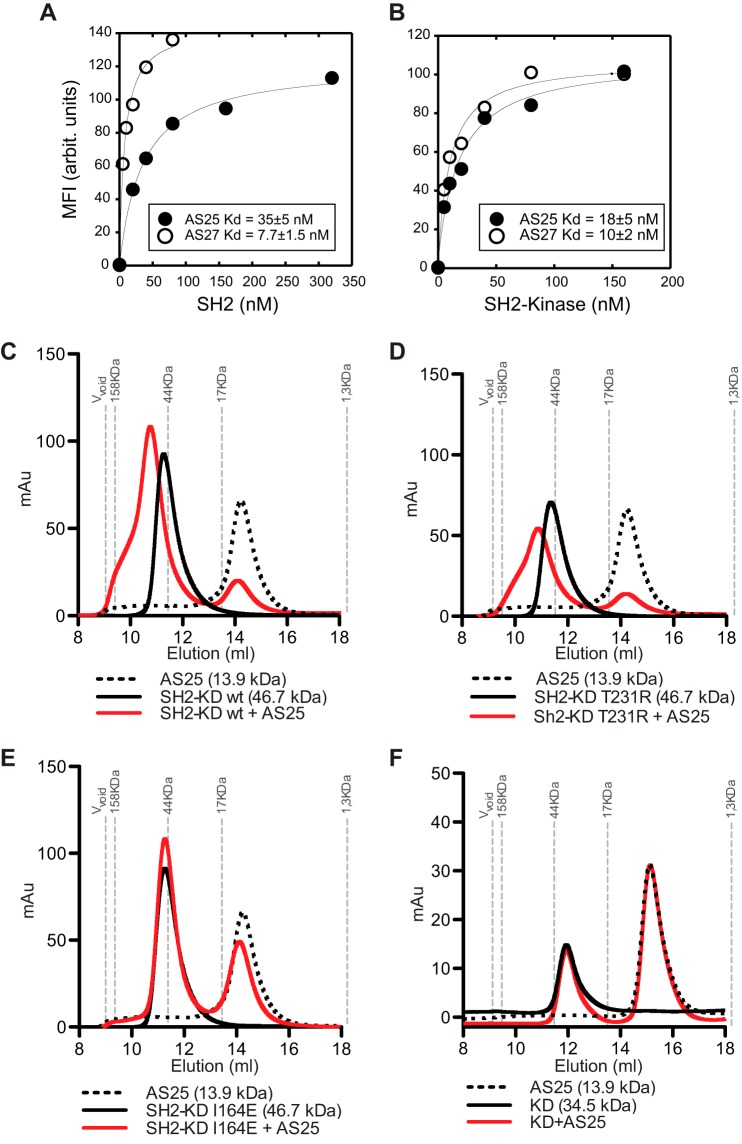
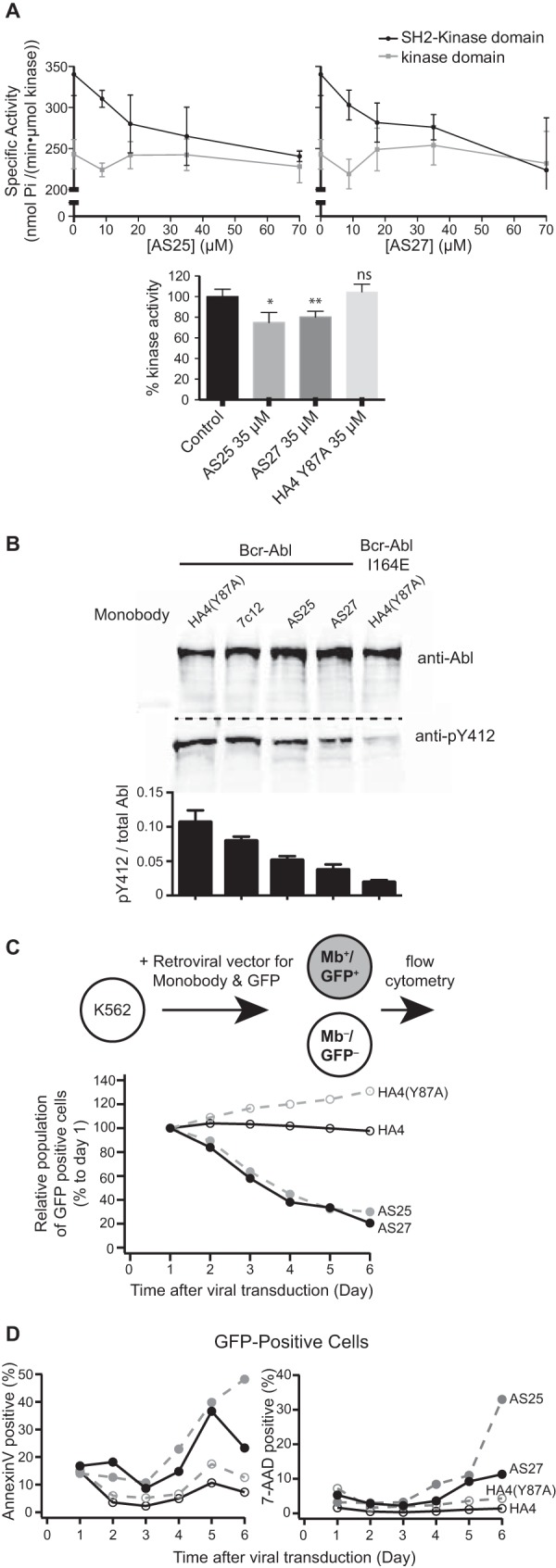
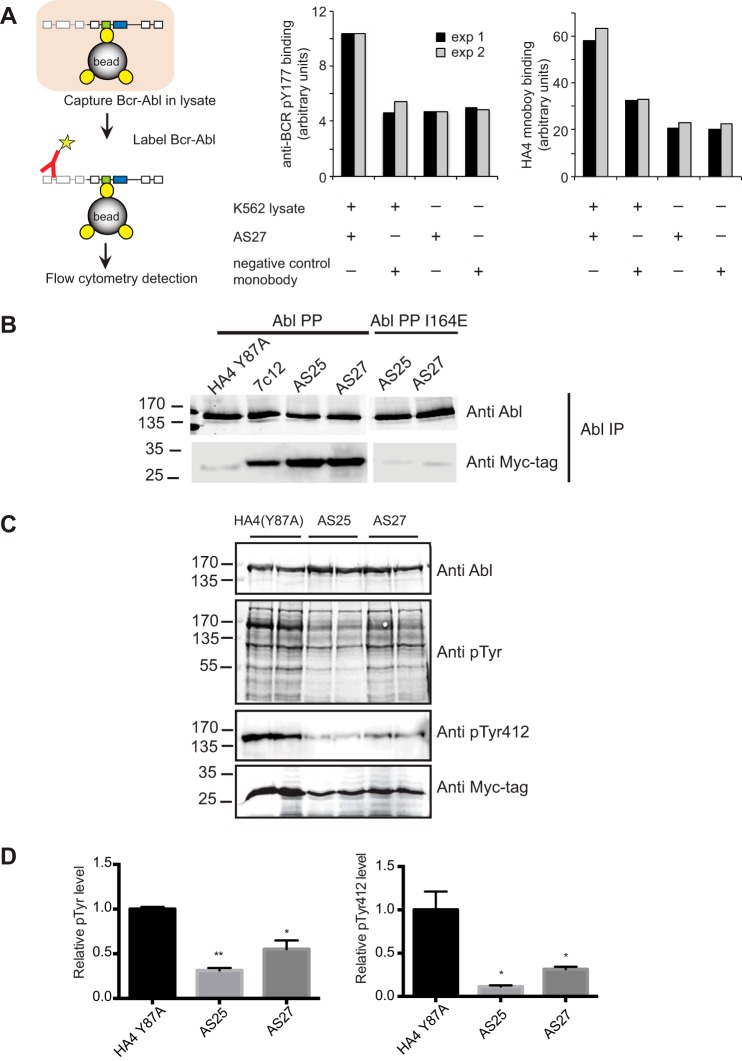
Bcr-Abl is a constitutively active kinase that causes chronic myelogenous leukemia. We have shown that a tandem fusion of two designed binding proteins, termed monobodies, directed to the interaction interface between the Src homology 2 (SH2) and kinase domains and to the phosphotyrosine-binding site of the SH2 domain, respectively, inhibits the Bcr-Abl kinase activity. Because the latter monobody inhibits processive phosphorylation by Bcr-Abl and the SH2-kinase interface is occluded in the active kinase, it remained undetermined whether targeting the SH2-kinase interface alone was sufficient for Bcr-Abl inhibition. To address this question, we generated new, higher affinity monobodies with single nanomolar KD values targeting the kinase-binding surface of SH2. Structural and mutagenesis studies revealed the molecular underpinnings of the monobody-SH2 interactions. Importantly, the new monobodies inhibited Bcr-Abl kinase activity in vitro and in cells, and they potently induced cell death in chronic myelogenous leukemia cell lines. This work provides strong evidence for the SH2-kinase interface as a pharmacologically tractable site for allosteric inhibition of Bcr-Abl.
Keywords: ABL tyrosine kinase; FN3; PPI inhibitor; Src homology 2 domain (SH2 domain); enzyme inhibitor; protein engineering; protein-protein interaction; x-ray crystallography.
© 2016 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures



References
-
- Holohan C., Van Schaeybroeck S., Longley D. B., and Johnston P. G. (2013) Cancer drug resistance: an evolving paradigm. Nat. Rev. Cancer 13, 714–726 - PubMed
-
- Shah N. P., and Sawyers C. L. (2003) Mechanisms of resistance to STI571 in Philadelphia chromosome-associated leukemias. Oncogene 22, 7389–7395 - PubMed
-
- Deininger M. W., Goldman J. M., and Melo J. V. (2000) The molecular biology of chronic myeloid leukemia. Blood 96, 3343–3356 - PubMed
-
- Shah N. P., Nicoll J. M., Nagar B., Gorre M. E., Paquette R. L., Kuriyan J., and Sawyers C. L. (2002) Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell 2, 117–125 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
