

Early Components of the Complement Classical Activation Pathway in Human Systemic Autoimmune Diseases
- PMID: 26913032
- PMCID: PMC4753731
- DOI: 10.3389/fimmu.2016.00036
Early Components of the Complement Classical Activation Pathway in Human Systemic Autoimmune Diseases
Abstract
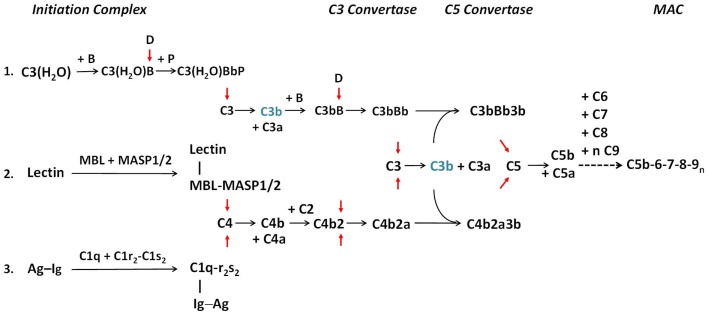
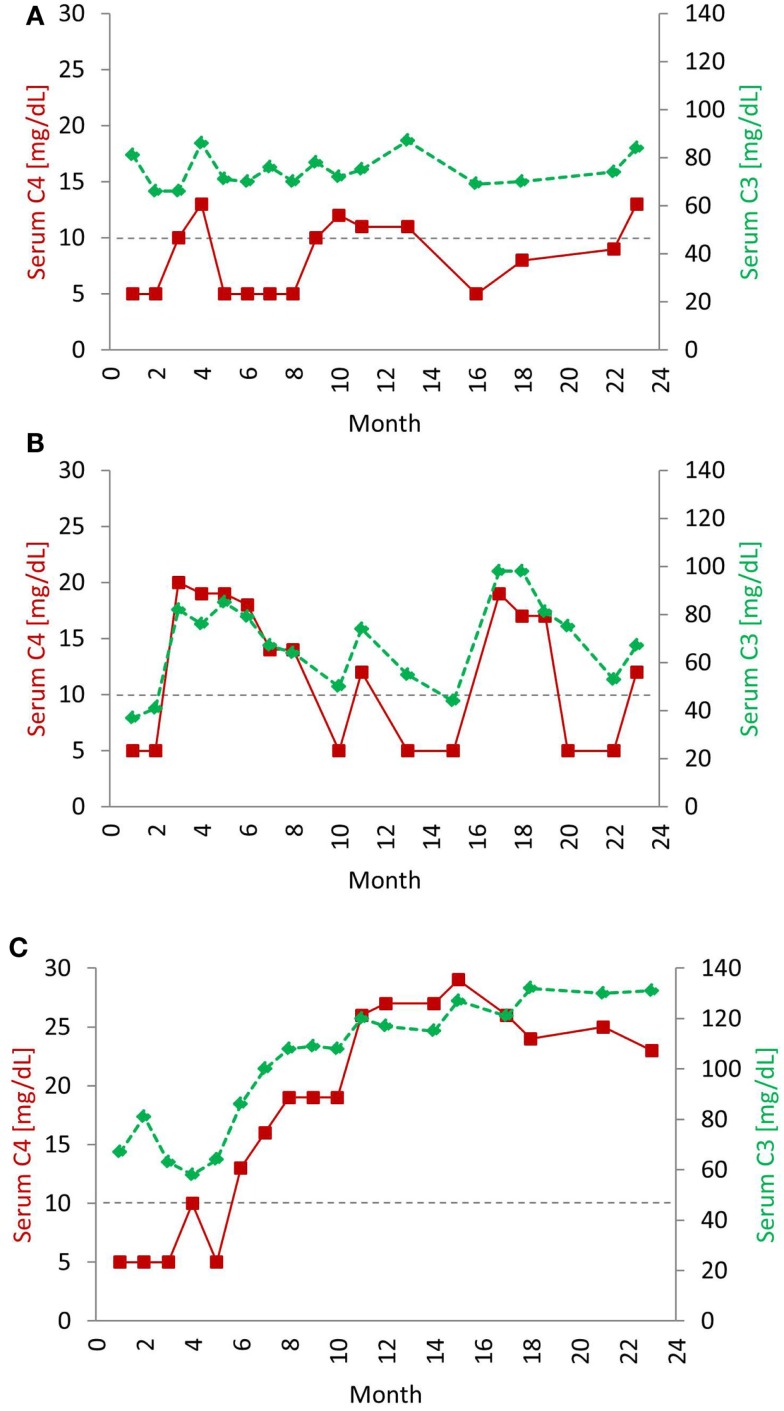
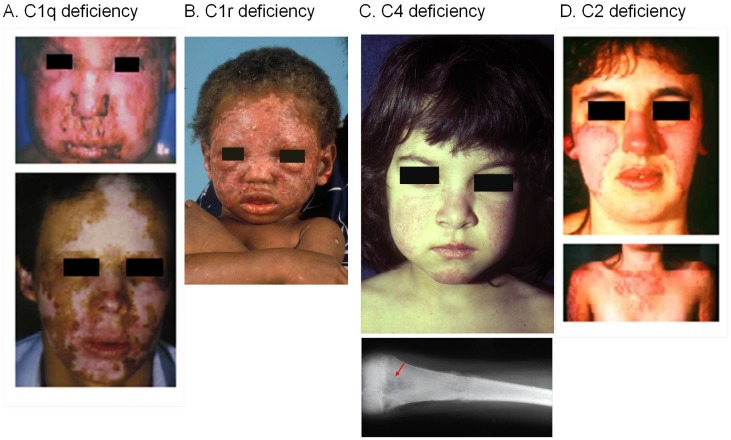
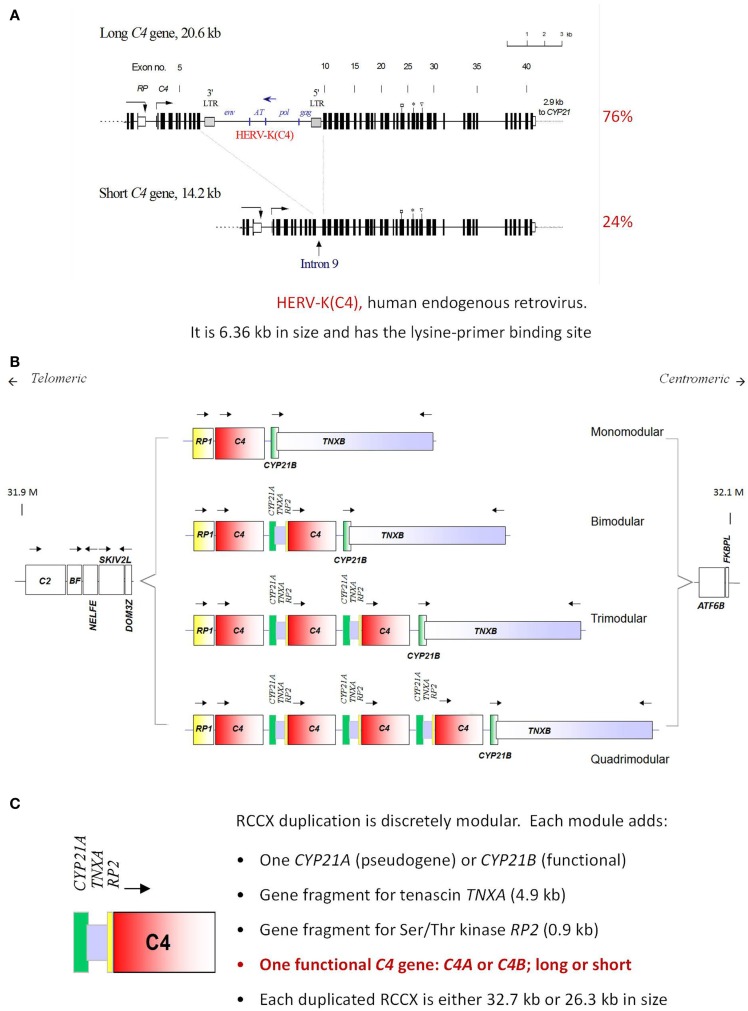
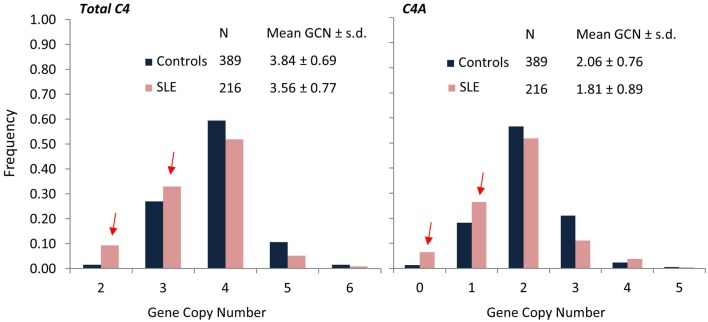
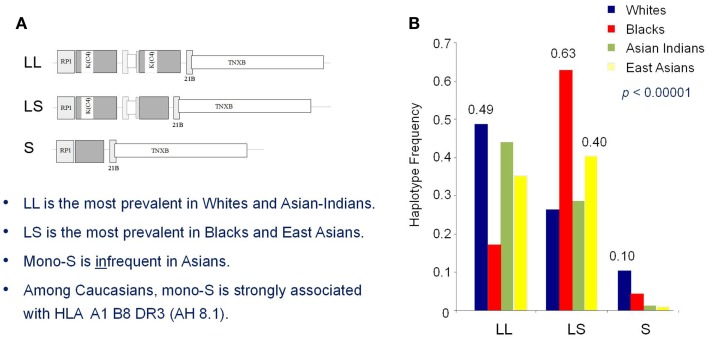
The complement system consists of effector proteins, regulators, and receptors that participate in host defense against pathogens. Activation of the complement system, via the classical pathway (CP), has long been recognized in immune complex-mediated tissue injury, most notably systemic lupus erythematosus (SLE). Paradoxically, a complete deficiency of an early component of the CP, as evidenced by homozygous genetic deficiencies reported in human, are strongly associated with the risk of developing SLE or a lupus-like disease. Similarly, isotype deficiency attributable to a gene copy-number (GCN) variation and/or the presence of autoantibodies directed against a CP component or a regulatory protein that result in an acquired deficiency are relatively common in SLE patients. Applying accurate assay methodologies with rigorous data validations, low GCNs of total C4, and heterozygous and homozygous deficiencies of C4A have been shown as medium to large effect size risk factors, while high copy numbers of total C4 or C4A as prevalent protective factors, of European and East-Asian SLE. Here, we summarize the current knowledge related to genetic deficiency and insufficiency, and acquired protein deficiencies for C1q, C1r, C1s, C4A/C4B, and C2 in disease pathogenesis and prognosis of SLE, and, briefly, for other systemic autoimmune diseases. As the complement system is increasingly found to be associated with autoimmune diseases and immune-mediated diseases, it has become an attractive therapeutic target. We highlight the recent developments and offer a balanced perspective concerning future investigations and therapeutic applications with a focus on early components of the CP in human systemic autoimmune diseases.
Keywords: autoimmune diseases; classical pathway; complement C1q; complement C1r; complement C1s; complement C2; complement C4; systemic lupus erythematosus.
Figures
References
-
- Yu C, Driest K, Wu Y, Lintner K, Patwardhan A, Spencer CH, et al. Complement in rheumatic diseases. In: Diamond B, Davidson A, editors. Encyclopedia of Medical Immunology: Autoimmune Diseases. New York, NY: Springer; (2014). p. 286–302.
-
- Atkinson J, Yu C. The complement system in systemic lupus erythematosus. In: Tsokos GC, editor. Systemic Lupus Erythematosus. Chapter 12, New York, NY: Elsevier; (2016). p. 81–112. 10.1016/B978-0-12-801917-7.00012-7 - DOI
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous