

EPG5-related Vici syndrome: a paradigm of neurodevelopmental disorders with defective autophagy
- PMID: 26917586
- PMCID: PMC4766378
- DOI: 10.1093/brain/awv393
EPG5-related Vici syndrome: a paradigm of neurodevelopmental disorders with defective autophagy
Abstract
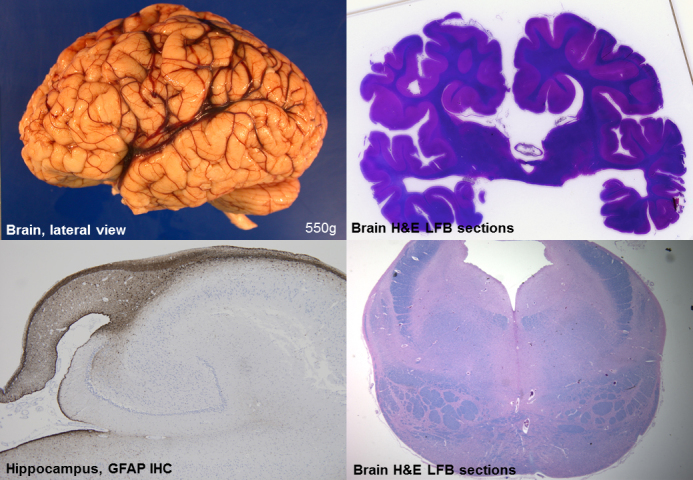
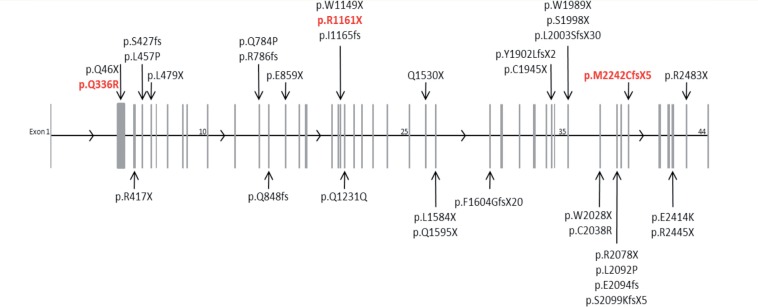
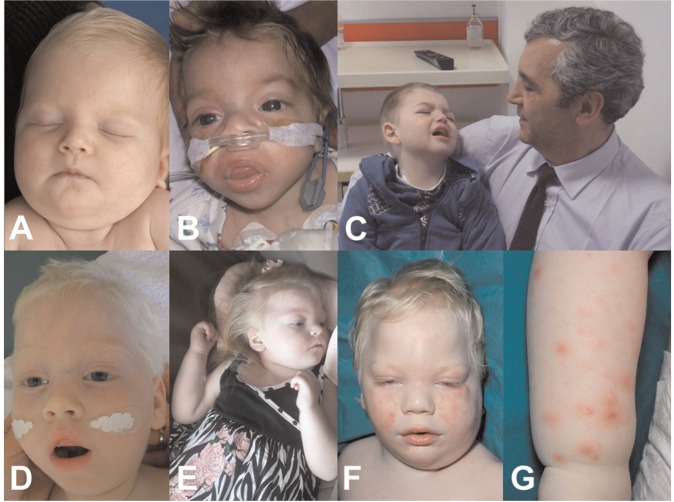
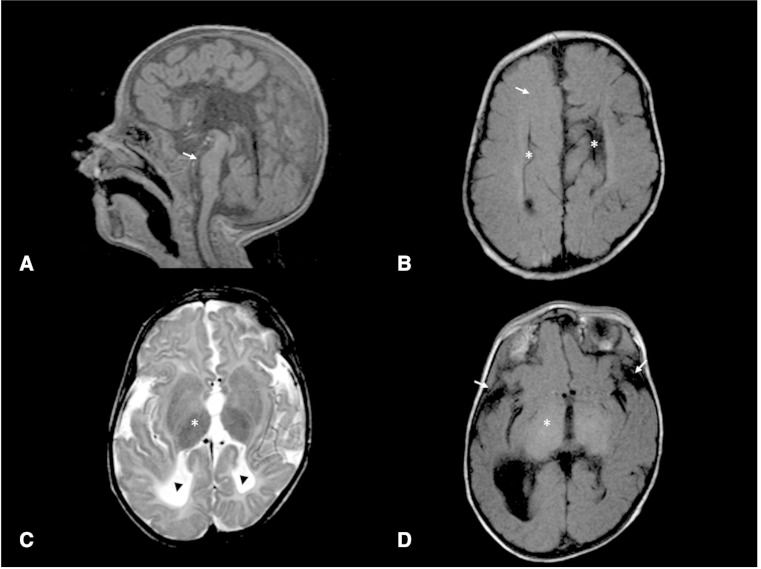
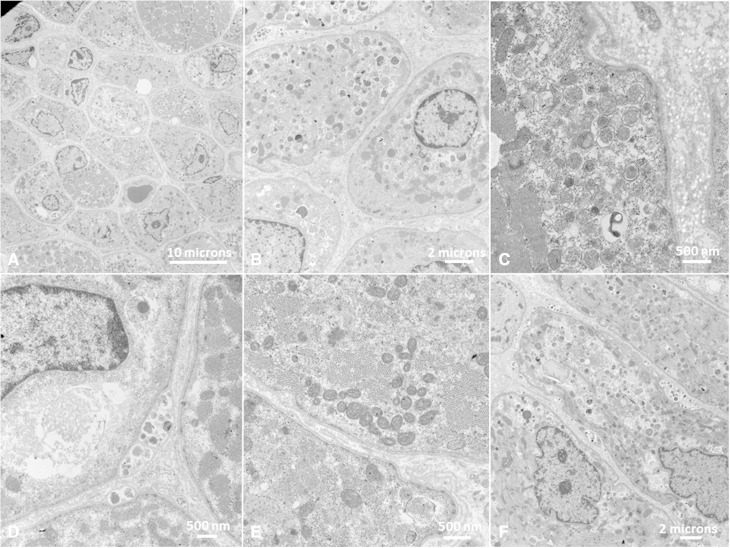
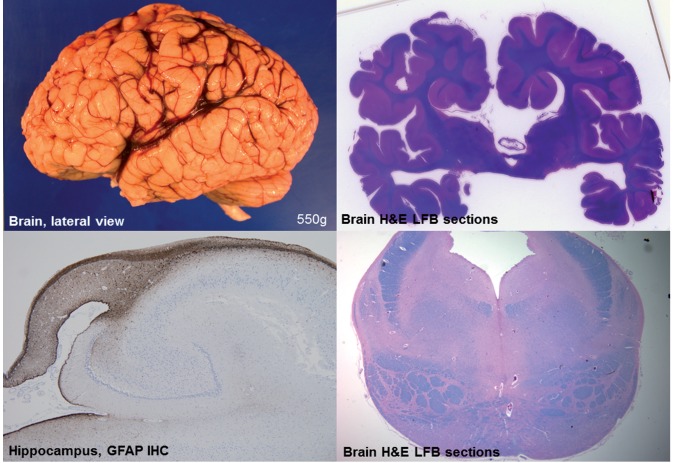
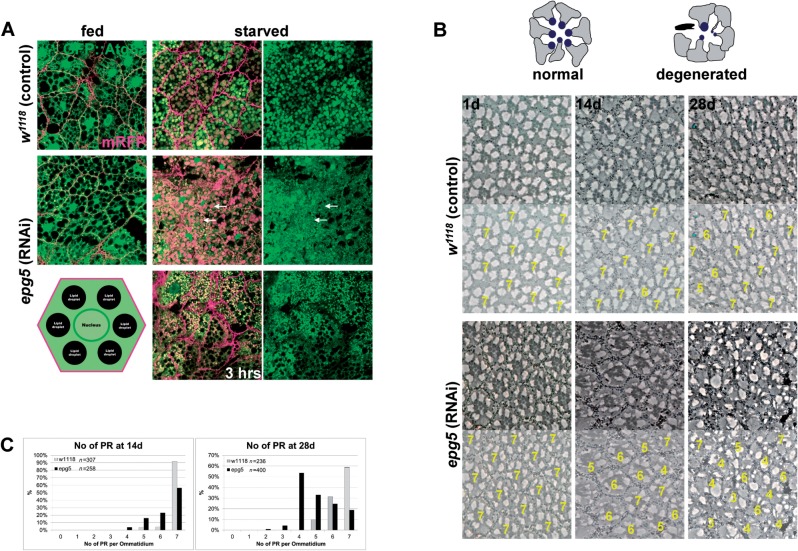
Vici syndrome is a progressive neurodevelopmental multisystem disorder due to recessive mutations in the key autophagy gene EPG5. We report genetic, clinical, neuroradiological, and neuropathological features of 50 children from 30 families, as well as the neuronal phenotype of EPG5 knock-down in Drosophila melanogaster. We identified 39 different EPG5 mutations, most of them truncating and predicted to result in reduced EPG5 protein. Most mutations were private, but three recurrent mutations (p.Met2242Cysfs*5, p.Arg417*, and p.Gln336Arg) indicated possible founder effects. Presentation was mainly neonatal, with marked hypotonia and feeding difficulties. In addition to the five principal features (callosal agenesis, cataracts, hypopigmentation, cardiomyopathy, and immune dysfunction), we identified three equally consistent features (profound developmental delay, progressive microcephaly, and failure to thrive). The manifestation of all eight of these features has a specificity of 97%, and a sensitivity of 89% for the presence of an EPG5 mutation and will allow informed decisions about genetic testing. Clinical progression was relentless and many children died in infancy. Survival analysis demonstrated a median survival time of 24 months (95% confidence interval 0-49 months), with only a 10th of patients surviving to 5 years of age. Survival outcomes were significantly better in patients with compound heterozygous mutations (P = 0.046), as well as in patients with the recurrent p.Gln336Arg mutation. Acquired microcephaly and regression of skills in long-term survivors suggests a neurodegenerative component superimposed on the principal neurodevelopmental defect. Two-thirds of patients had a severe seizure disorder, placing EPG5 within the rapidly expanding group of genes associated with early-onset epileptic encephalopathies. Consistent neuroradiological features comprised structural abnormalities, in particular callosal agenesis and pontine hypoplasia, delayed myelination and, less frequently, thalamic signal intensity changes evolving over time. Typical muscle biopsy features included fibre size variability, central/internal nuclei, abnormal glycogen storage, presence of autophagic vacuoles and secondary mitochondrial abnormalities. Nerve biopsy performed in one case revealed subtotal absence of myelinated axons. Post-mortem examinations in three patients confirmed neurodevelopmental and neurodegenerative features and multisystem involvement. Finally, downregulation of epg5 (CG14299) in Drosophila resulted in autophagic abnormalities and progressive neurodegeneration. We conclude that EPG5-related Vici syndrome defines a novel group of neurodevelopmental disorders that should be considered in patients with suggestive features in whom mitochondrial, glycogen, or lysosomal storage disorders have been excluded. Neurological progression over time indicates an intriguing link between neurodevelopment and neurodegeneration, also supported by neurodegenerative features in epg5-deficient Drosophila, and recent implication of other autophagy regulators in late-onset neurodegenerative disease.
Keywords: EPG5; Vici syndrome; callosal agenesis; ectopic P granules autophagy protein 5; neurodegeneration; neurodevelopment.
© The Author (2016). Published by Oxford University Press on behalf of the Guarantors of Brain.
Figures
Comment in
-
Aberrant splicing induced by the most common EPG5 mutation in an individual with Vici syndrome.Brain. 2016 Sep;139(Pt 9):e52. doi: 10.1093/brain/aww135. Epub 2016 Jun 24. Brain. 2016. PMID: 27343256 Free PMC article. No abstract available.
-
Reply: Aberrant splicing induced by the most common EPG5 mutation in an individual with Vici syndrome.Brain. 2016 Sep;139(Pt 9):e53. doi: 10.1093/brain/aww136. Epub 2016 Jun 24. Brain. 2016. PMID: 27343258 No abstract available.
References
-
- Al-Owain M, Al-Hashem A, Al-Muhaizea M, Humaidan H, Al-Hindi H, Al-Homoud I, et al. Vici syndrome associated with unilateral lung hypoplasia and myopathy. Am J Med Genet A 2010; 152: 1849–53. - PubMed
-
- Autti T, Joensuu R, Aberg L. Decreased T2 signal in the thalami may be a sign of lysosomal storage disease. Neuroradiology 2007; 49: 571–8. - PubMed
-
- Brockmann K, Berg D. The significance of GBA for Parkinson's disease. J Inherit Metab Dis 2014; 37: 643–8. - PubMed
-
- Byrne S, Dlamini N, Lumsden D, Pitt M, Zaharieva I, Muntoni F, et al. SIL1-related Marinesco-Sjoegren syndrome (MSS) with associated motor neuronopathy and bradykinetic movement disorder. Neuromuscul Disord 2015; 25: 585–8. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
