

Drug-Induced Metabolic Acidosis
- PMID: 26918138
- PMCID: PMC4754009
- DOI: 10.12688/f1000research.7006.1
Drug-Induced Metabolic Acidosis
Abstract
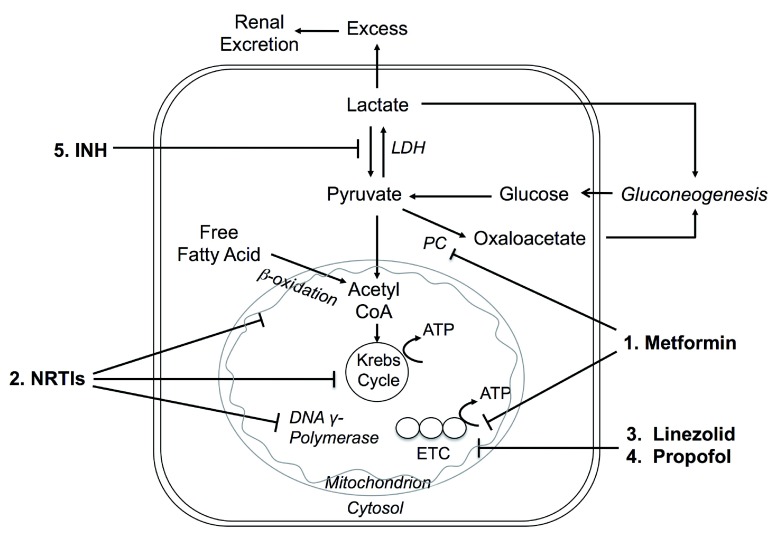
Metabolic acidosis could emerge from diseases disrupting acid-base equilibrium or from drugs that induce similar derangements. Occurrences are usually accompanied by comorbid conditions of drug-induced metabolic acidosis, and clinical outcomes may range from mild to fatal. It is imperative that clinicians not only are fully aware of the list of drugs that may lead to metabolic acidosis but also understand the underlying pathogenic mechanisms. In this review, we categorized drug-induced metabolic acidosis in terms of pathophysiological mechanisms, as well as individual drugs' characteristics.
Keywords: MALA; acidosis; drug-induced; metabolic.
Conflict of interest statement
No competing interests were disclosed.
Figures




References
-
- Moe OW, Fuster D, Alpern RJ: Common acid-base disorders. In: Goldman L, Wachter RM, Hollander H, editors. Hosp Med 2nd ed. Philadelphia: Lippincott, William & Wilkins;2005;1055–65.
-
- Wiederkehr MR, Moe OW: Treatment of metabolic acidosis. In: Massry SG, Suki WK, editors. Therapy of Renal Diseases and Related Disorders 4th ed: Springer; In press,2011.
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical