

NF-κB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria
- PMID: 26919428
- PMCID: PMC4769378
- DOI: 10.1016/j.cell.2015.12.057
NF-κB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria
Abstract
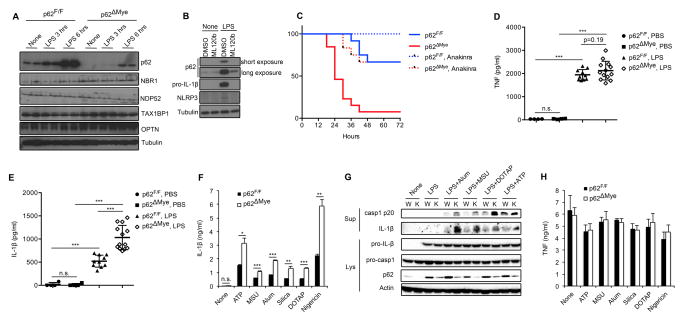
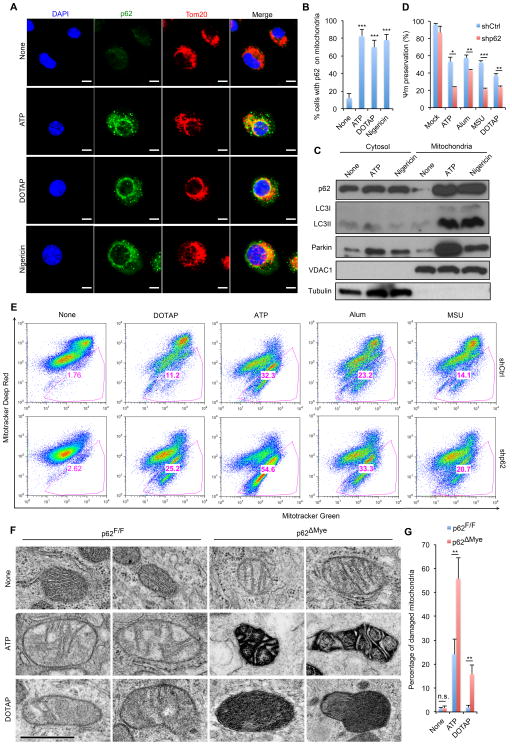
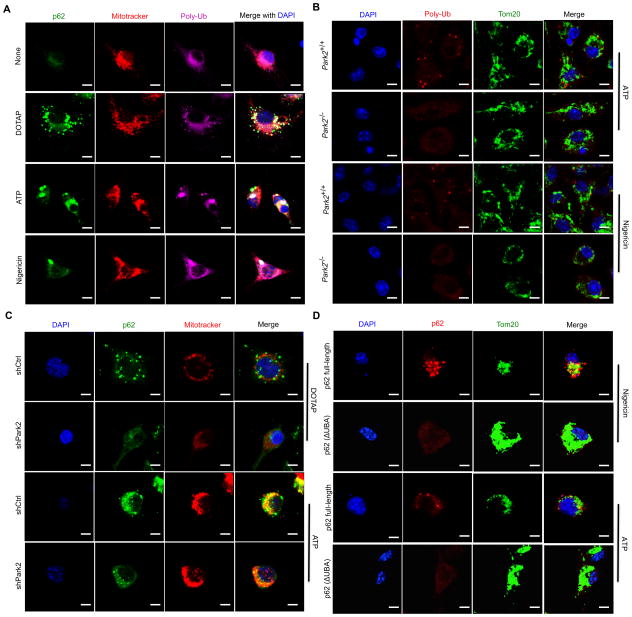
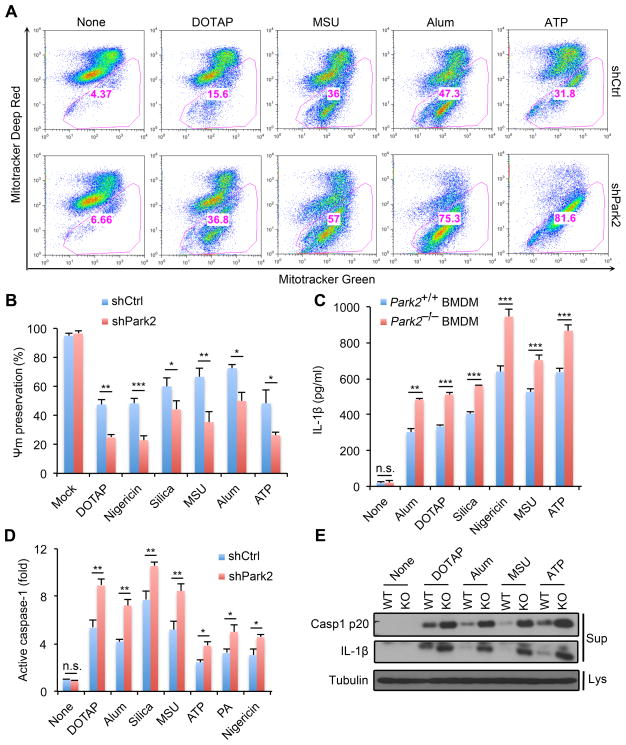
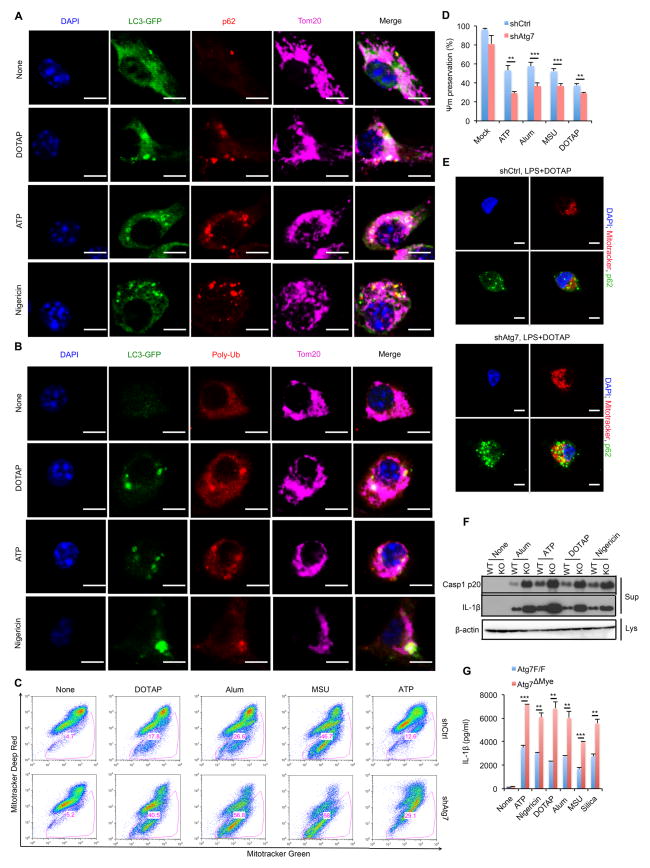
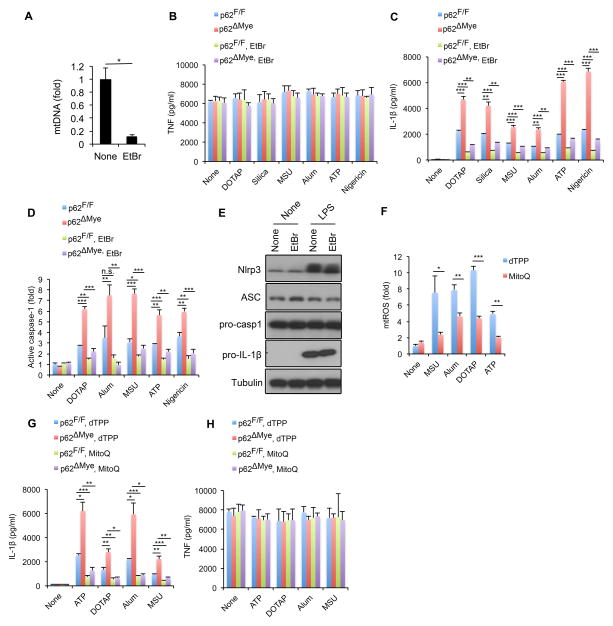
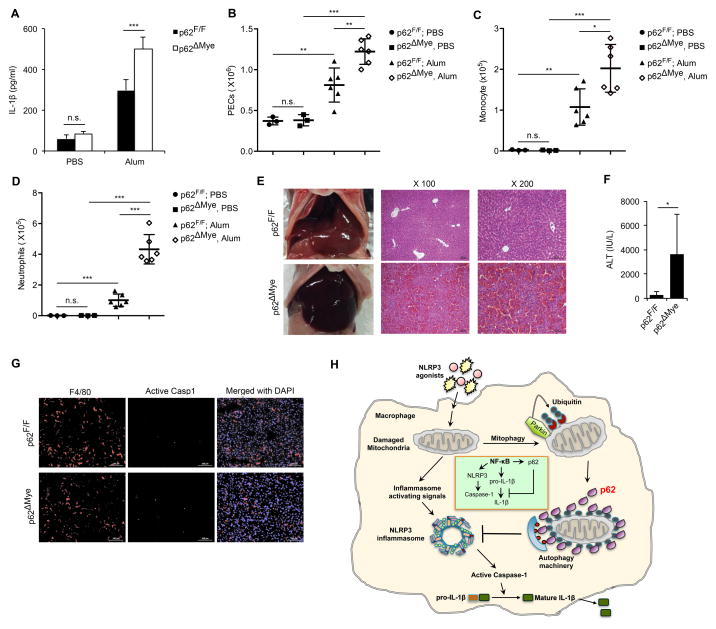
Nuclear factor κB (NF-κB), a key activator of inflammation, primes the NLRP3-inflammasome for activation by inducing pro-IL-1β and NLRP3 expression. NF-κB, however, also prevents excessive inflammation and restrains NLRP3-inflammasome activation through a poorly defined mechanism. We now show that NF-κB exerts its anti-inflammatory activity by inducing delayed accumulation of the autophagy receptor p62/SQSTM1. External NLRP3-activating stimuli trigger a form of mitochondrial (mt) damage that is caspase-1- and NLRP3-independent and causes release of direct NLRP3-inflammasome activators, including mtDNA and mtROS. Damaged mitochondria undergo Parkin-dependent ubiquitin conjugation and are specifically recognized by p62, which induces their mitophagic clearance. Macrophage-specific p62 ablation causes pronounced accumulation of damaged mitochondria and excessive IL-1β-dependent inflammation, enhancing macrophage death. Therefore, the "NF-κB-p62-mitophagy" pathway is a macrophage-intrinsic regulatory loop through which NF-κB restrains its own inflammation-promoting activity and orchestrates a self-limiting host response that maintains homeostasis and favors tissue repair.
Copyright © 2016 Elsevier Inc. All rights reserved.
Figures
Comment in
-
Clean Up after Yourself.Mol Cell. 2016 Mar 3;61(5):644-645. doi: 10.1016/j.molcel.2016.02.021. Mol Cell. 2016. PMID: 26942667
-
Inflammasome: Anti-inflammatory effect of mitophagy.Nat Rev Immunol. 2016 Apr;16(4):206. doi: 10.1038/nri.2016.33. Epub 2016 Mar 14. Nat Rev Immunol. 2016. PMID: 26972724 No abstract available.
References
-
- Barnes PJ, Karin M. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997;336:1066–1071. - PubMed
-
- Baroja-Mazo A, Martin-Sanchez F, Gomez AI, Martinez CM, Amores-Iniesta J, Compan V, Barbera-Cremades M, Yague J, Ruiz-Ortiz E, Anton J, et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat Immunol. 2014;15:738–748. - PubMed
-
- Brenner C, Galluzzi L, Kepp O, Kroemer G. Decoding cell death signals in liver inflammation. J Hepatol. 2013;59:583–594. - PubMed
-
- Chernyak BV, Izyumov DS, Lyamzaev KG, Pashkovskaya AA, Pletjushkina OY, Antonenko YN, Sakharov DV, Wirtz KW, Skulachev VP. Production of reactive oxygen species in mitochondria of HeLa cells under oxidative stress. Biochim Biophys Acta. 2006;1757:525–534. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 AI052430/AI/NIAID NIH HHS/United States
- AI043477/AI/NIAID NIH HHS/United States
- P01 CA128814/CA/NCI NIH HHS/United States
- CA030199/CA/NCI NIH HHS/United States
- R01 CA172025/CA/NCI NIH HHS/United States
- CA163798/CA/NCI NIH HHS/United States
- AI52430/AI/NIAID NIH HHS/United States
- AA020172/AA/NIAAA NIH HHS/United States
- MFE-135425/CAPMC/ CIHR/Canada
- CA192642/CA/NCI NIH HHS/United States
- R01 CA132847/CA/NCI NIH HHS/United States
- R01 CA155120/CA/NCI NIH HHS/United States
- R01 CA192642/CA/NCI NIH HHS/United States
- R01 HL087023/HL/NHLBI NIH HHS/United States
- P41 GM103412/GM/NIGMS NIH HHS/United States
- DK085252/DK/NIDDK NIH HHS/United States
- R37 AI043477/AI/NIAID NIH HHS/United States
- R01 AA020172/AA/NIAAA NIH HHS/United States
- P01 DK098108/DK/NIDDK NIH HHS/United States
- GM103412/GM/NIGMS NIH HHS/United States
- P42 ES010337/ES/NIEHS NIH HHS/United States
- P30 CA030199/CA/NCI NIH HHS/United States
- R01 DK085252/DK/NIDDK NIH HHS/United States
- CA172025/CA/NCI NIH HHS/United States
- HL087023/HL/NHLBI NIH HHS/United States
- ES010337/ES/NIEHS NIH HHS/United States
- CA132847/CA/NCI NIH HHS/United States
- R01 CA163798/CA/NCI NIH HHS/United States
- R01 AI043477/AI/NIAID NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
