

Linking Pathogenic Mechanisms of Alcoholic Liver Disease With Clinical Phenotypes
- PMID: 26919968
- PMCID: PMC4887335
- DOI: 10.1053/j.gastro.2016.02.035
Linking Pathogenic Mechanisms of Alcoholic Liver Disease With Clinical Phenotypes
Abstract
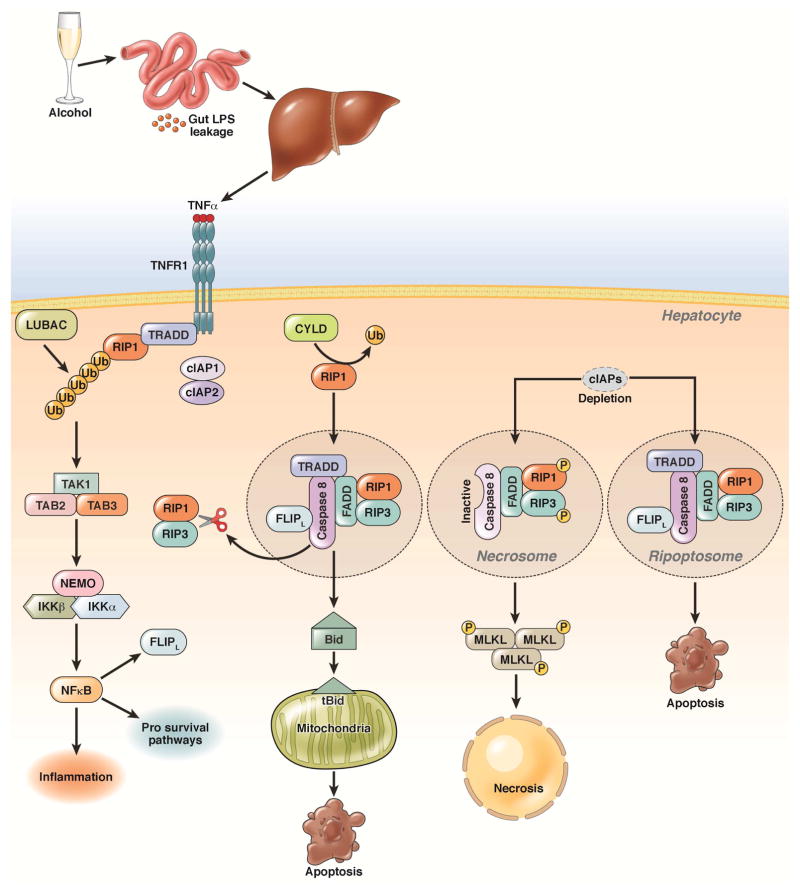
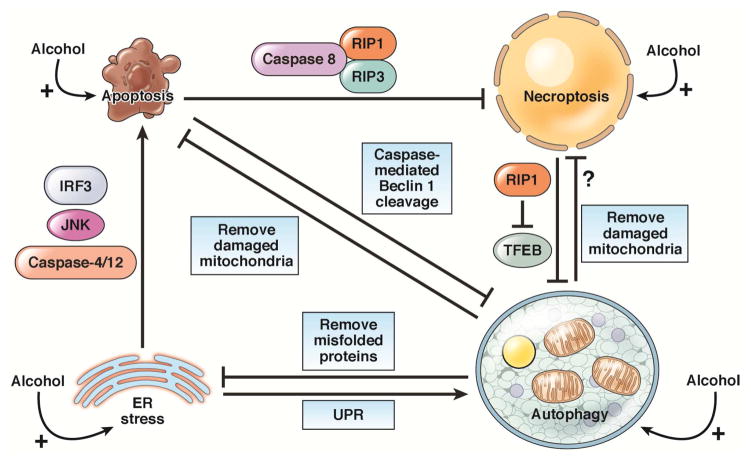
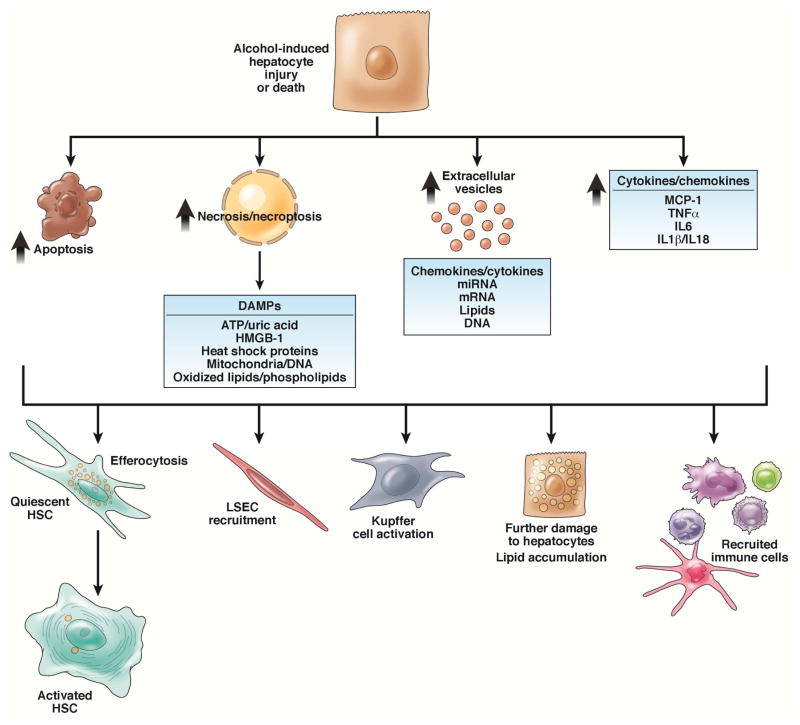
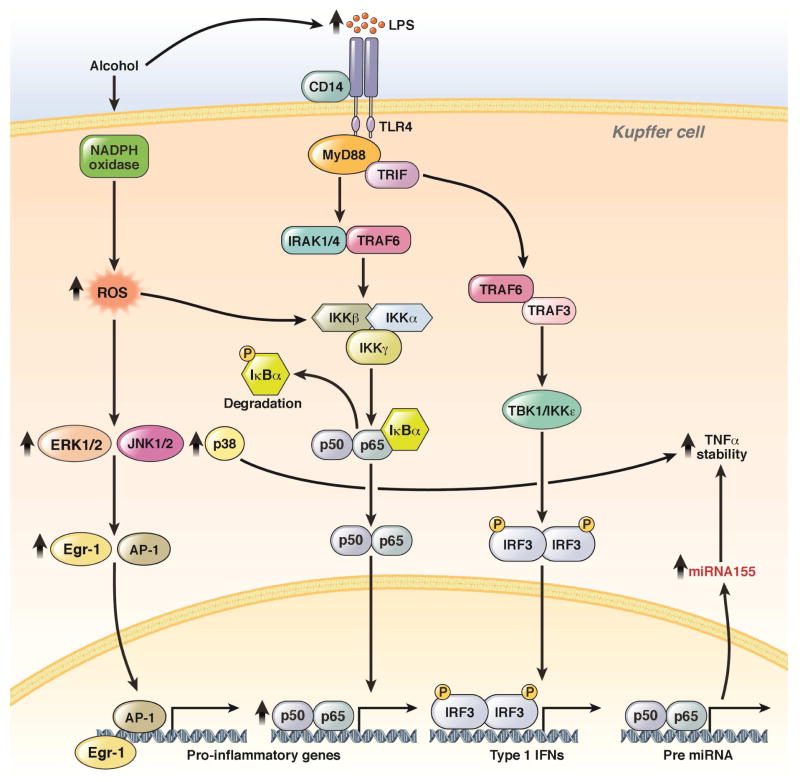
Alcoholic liver disease (ALD) develops in approximately 20% of alcoholic patients, with a higher prevalence in females. ALD progression is marked by fatty liver and hepatocyte necrosis, as well as apoptosis, inflammation, regenerating nodules, fibrosis, and cirrhosis.(1) ALD develops via a complex process involving parenchymal and nonparenchymal cells, as well as recruitment of other cell types to the liver in response to damage and inflammation. Hepatocytes are damaged by ethanol, via generation of reactive oxygen species and induction of endoplasmic reticulum stress and mitochondrial dysfunction. Hepatocyte cell death via apoptosis and necrosis are markers of ethanol-induced liver injury. We review the mechanisms by which alcohol injures hepatocytes and the response of hepatic sinusoidal cells to alcohol-induced injury. We also discuss how recent insights into the pathogenesis of ALD will affect the treatment and management of patients.
Keywords: Alcoholic Hepatitis; Alcoholic Liver Disease; Hepatic Stellate Cell.
Copyright © 2016 AGA Institute. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Figures
References
-
- Crabb DW, Matsumoto M, Chang D, et al. Overview of the role of alcohol dehydrogenase and aldehyde dehydrogenase and their variants in the genesis of alcohol-related pathology. Proc Nutr Soc. 2004;63:49–63. - PubMed
-
- Cao Q, Mak KM, Lieber CS. Cytochrome P4502E1 primes macrophages to increase TNF-alpha production in response to lipopolysaccharide. Am J Physiol Gastrointest Liver Physiol. 2005;289:G95–107. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
