

Exploiting biological priors and sequence variants enhances QTL discovery and genomic prediction of complex traits
- PMID: 26920147
- PMCID: PMC4769584
- DOI: 10.1186/s12864-016-2443-6
Exploiting biological priors and sequence variants enhances QTL discovery and genomic prediction of complex traits
Abstract
Background: Dense SNP genotypes are often combined with complex trait phenotypes to map causal variants, study genetic architecture and provide genomic predictions for individuals with genotypes but no phenotype. A single method of analysis that jointly fits all genotypes in a Bayesian mixture model (BayesR) has been shown to competitively address all 3 purposes simultaneously. However, BayesR and other similar methods ignore prior biological knowledge and assume all genotypes are equally likely to affect the trait. While this assumption is reasonable for SNP array genotypes, it is less sensible if genotypes are whole-genome sequence variants which should include causal variants.
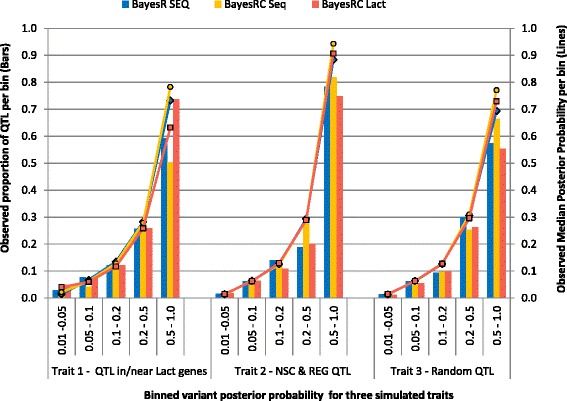
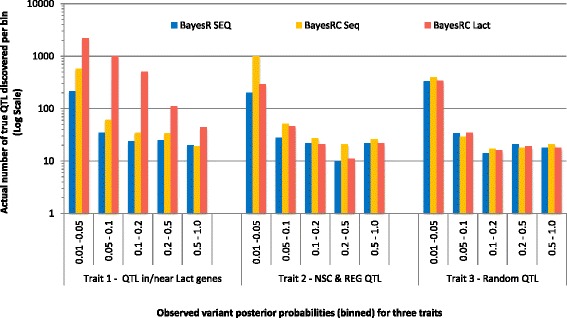
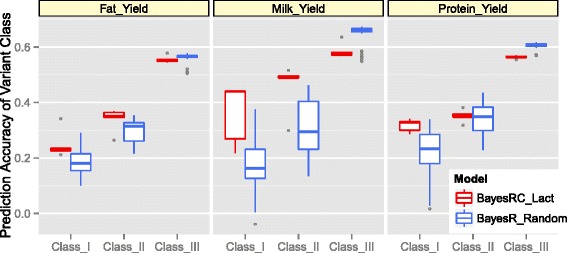
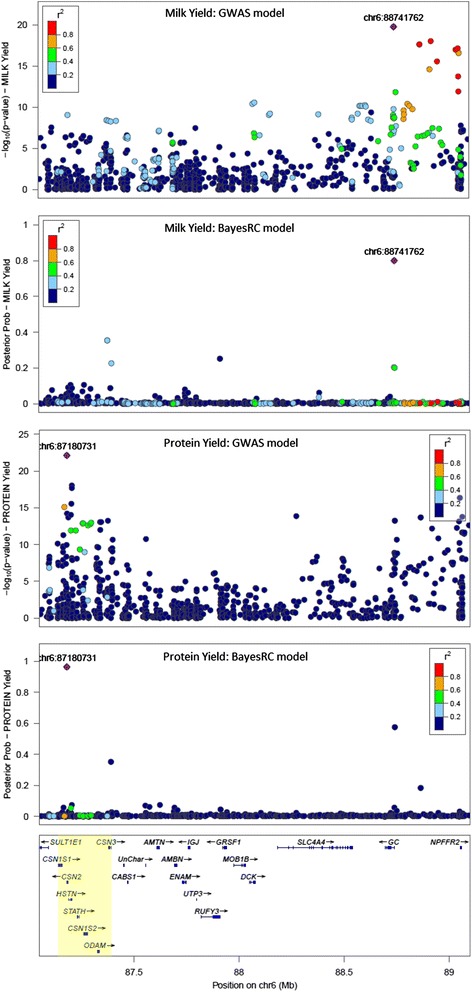
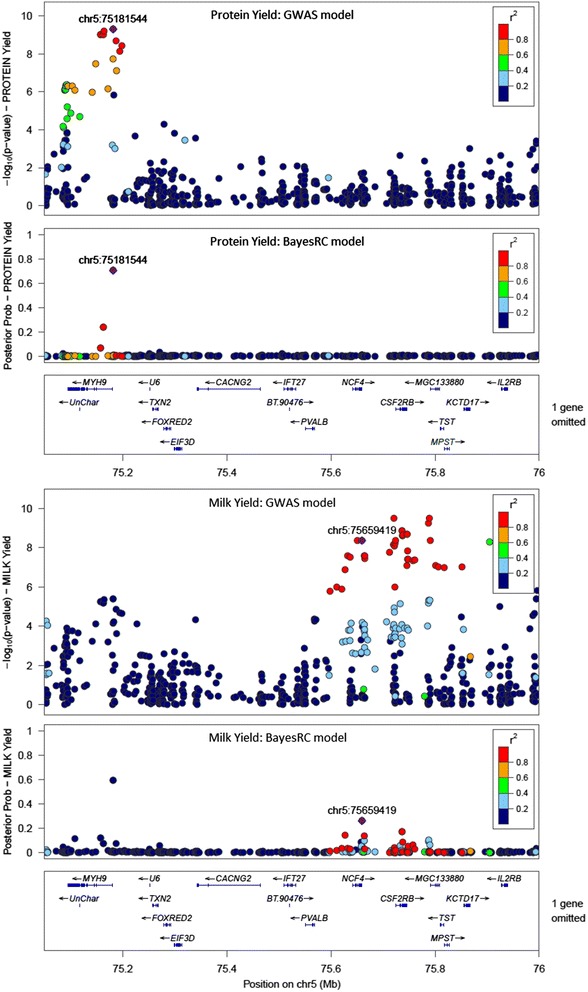
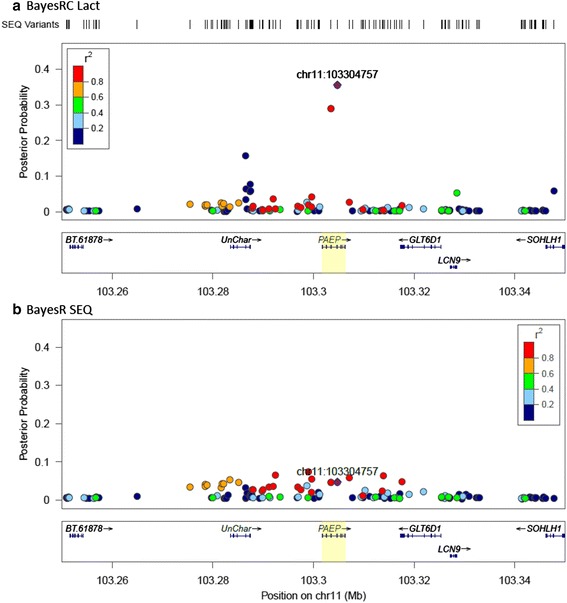
Results: We introduce a new method (BayesRC) based on BayesR that incorporates prior biological information in the analysis by defining classes of variants likely to be enriched for causal mutations. The information can be derived from a range of sources, including variant annotation, candidate gene lists and known causal variants. This information is then incorporated objectively in the analysis based on evidence of enrichment in the data. We demonstrate the increased power of BayesRC compared to BayesR using real dairy cattle genotypes with simulated phenotypes. The genotypes were imputed whole-genome sequence variants in coding regions combined with dense SNP markers. BayesRC increased the power to detect causal variants and increased the accuracy of genomic prediction. The relative improvement for genomic prediction was most apparent in validation populations that were not closely related to the reference population. We also applied BayesRC to real milk production phenotypes in dairy cattle using independent biological priors from gene expression analyses. Although current biological knowledge of which genes and variants affect milk production is still very incomplete, our results suggest that the new BayesRC method was equal to or more powerful than BayesR for detecting candidate causal variants and for genomic prediction of milk traits.
Conclusions: BayesRC provides a novel and flexible approach to simultaneously improving the accuracy of QTL discovery and genomic prediction by taking advantage of prior biological knowledge. Approaches such as BayesRC will become increasing useful as biological knowledge accumulates regarding functional regions of the genome for a range of traits and species.
Figures

References
-
- Kemper KE, Reich CM, Bowman P, vander Jagt CJ, Chamberlain AJ, Mason BA, et al. Improved precision of QTL mapping using a nonlinear Bayesian method in a multi-breed population leads to greater accuracy of across-breed genomic predictions. Genet Sel Evol. 2015;47(1):29. doi: 10.1186/s12711-014-0074-4. - DOI - PMC - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
