

Germline and somatic FGFR1 abnormalities in dysembryoplastic neuroepithelial tumors
- PMID: 26920151
- PMCID: PMC5039033
- DOI: 10.1007/s00401-016-1549-x
Germline and somatic FGFR1 abnormalities in dysembryoplastic neuroepithelial tumors
Abstract
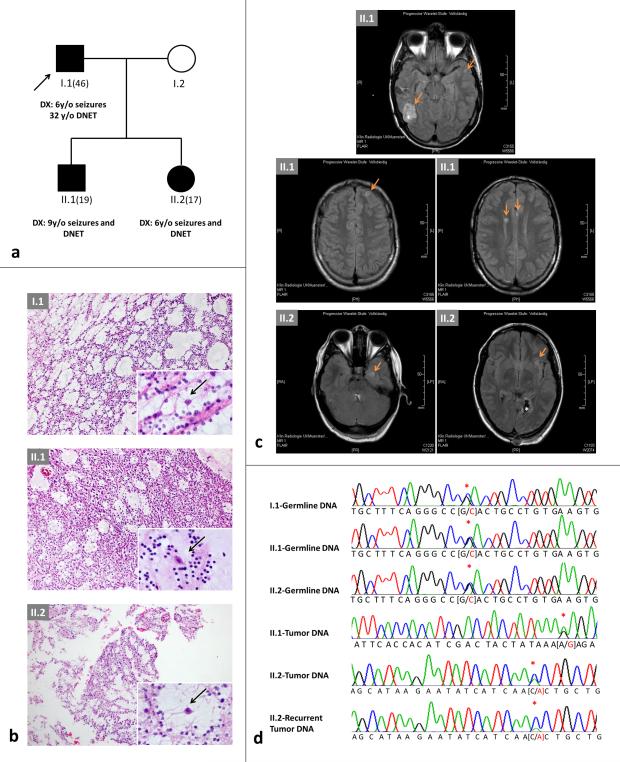
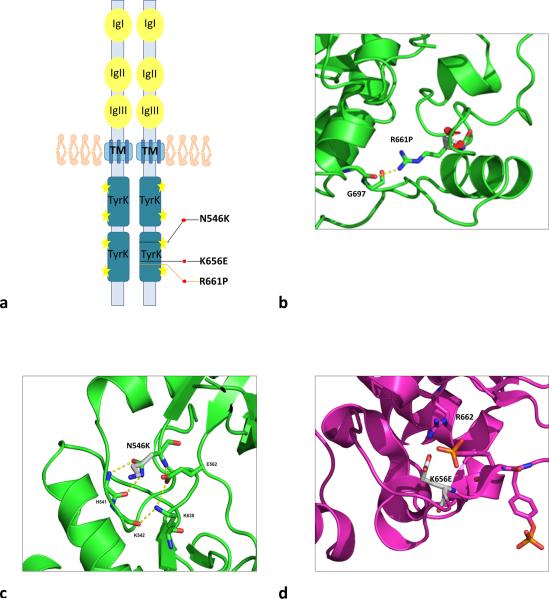
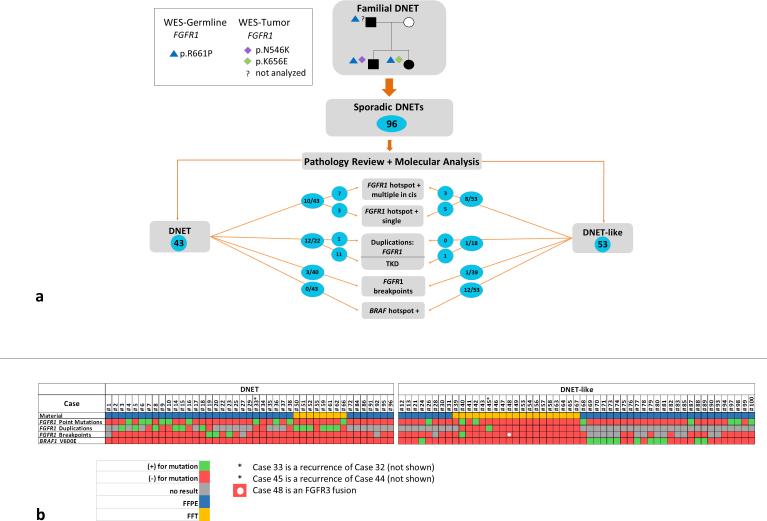
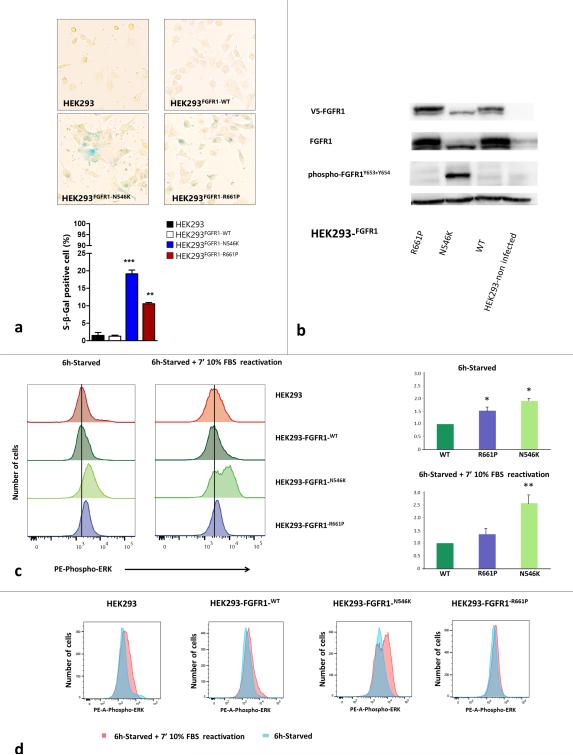
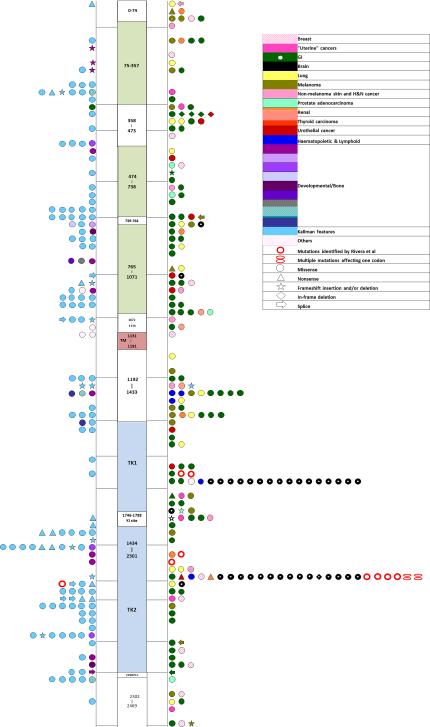
Dysembryoplastic neuroepithelial tumor (DNET) is a benign brain tumor associated with intractable drug-resistant epilepsy. In order to identify underlying genetic alterations and molecular mechanisms, we examined three family members affected by multinodular DNETs as well as 100 sporadic tumors from 96 patients, which had been referred to us as DNETs. We performed whole-exome sequencing on 46 tumors and targeted sequencing for hotspot FGFR1 mutations and BRAF p.V600E was used on the remaining samples. FISH, copy number variation assays and Sanger sequencing were used to validate the findings. By whole-exome sequencing of the familial cases, we identified a novel germline FGFR1 mutation, p.R661P. Somatic activating FGFR1 mutations (p.N546K or p.K656E) were observed in the tumor samples and further evidence for functional relevance was obtained by in silico modeling. The FGFR1 p.K656E mutation was confirmed to be in cis with the germline p.R661P variant. In 43 sporadic cases, in which the diagnosis of DNET could be confirmed on central blinded neuropathology review, FGFR1 alterations were also frequent and mainly comprised intragenic tyrosine kinase FGFR1 duplication and multiple mutants in cis (25/43; 58.1 %) while BRAF p.V600E alterations were absent (0/43). In contrast, in 53 cases, in which the diagnosis of DNET was not confirmed, FGFR1 alterations were less common (10/53; 19 %; p < 0.0001) and hotspot BRAF p.V600E (12/53; 22.6 %) (p < 0.001) prevailed. We observed overexpression of phospho-ERK in FGFR1 p.R661P and p.N546K mutant expressing HEK293 cells as well as FGFR1 mutated tumor samples, supporting enhanced MAP kinase pathway activation under these conditions. In conclusion, constitutional and somatic FGFR1 alterations and MAP kinase pathway activation are key events in the pathogenesis of DNET. These findings point the way towards existing targeted therapies.
Keywords: Brain development; Epilepsy; FGFR signaling; Molecular etiology; Targeted therapy; Whole-exome sequencing.
Figures
References
-
- Barba C, Jacques T, Kahane P, Polster T, Isnard J, Leijten FS, Ozkara C, Tassi L, Giordano F, Castagna M, John A, Oz B, Salon C, Streichenberger N, Cross JH, Guerrini R. Epilepsy surgery in Neurofibromatosis Type 1. Epilepsy Res. 2013;105:384–395. doi:10.1016/j.eplepsyres.2013.02.021. - PubMed
-
- Blümcke I, Aronica E, Urbach H, Alexopoulos A, Gonzalez-Martinez JA. A neuropathology-based approach to epilepsy surgery in brain tumors and proposal for a new terminology use for long-term epilepsy-associated brain tumors. Acta Neuropathol. 2014;128:39–54. doi:10.1007/s00401-014-1288-9. - PMC - PubMed
-
- Chappe C, Padovani L, Scavarda D, Forest F, Nanni-Metellus I, Loundou A, Mercurio S, Fina F, Lena G, Colin C, Figarella-Branger D. Dysembryoplastic neuroepithelial tumors share with pleomorphic xanthoastrocytomas and gangliogliomas BRAF(V600E) mutation and expression. Brain Pathol. 2013;23:574–583. doi:10.1111/bpa.12048. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
