

Case report of novel DYRK1A mutations in 2 individuals with syndromic intellectual disability and a review of the literature
- PMID: 26922654
- PMCID: PMC4769499
- DOI: 10.1186/s12881-016-0276-4
Case report of novel DYRK1A mutations in 2 individuals with syndromic intellectual disability and a review of the literature
Abstract
Background: Chromosomal deletions encompassing DYRK1A have been associated with intellectual disability for several years. More recently, point mutations in DYRK1A have been shown to be responsible for a recognizable syndrome characterized by microcephaly, developmental delay and intellectual disability (ID) as well as characteristic facial features. Here we present 2 individuals with novel mutations in DYRK1A, and a review of the cases reported to date.
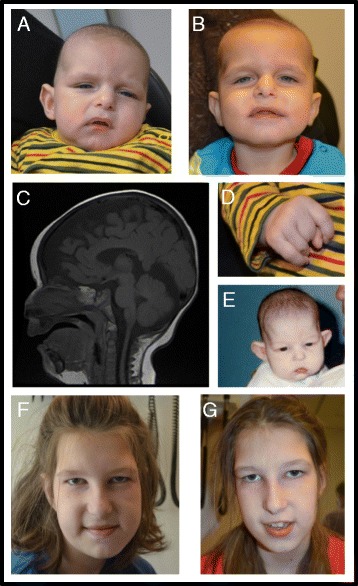
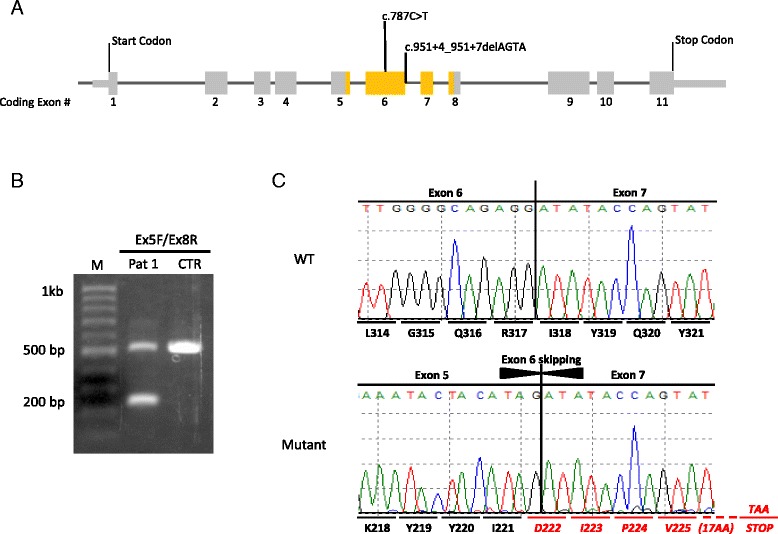
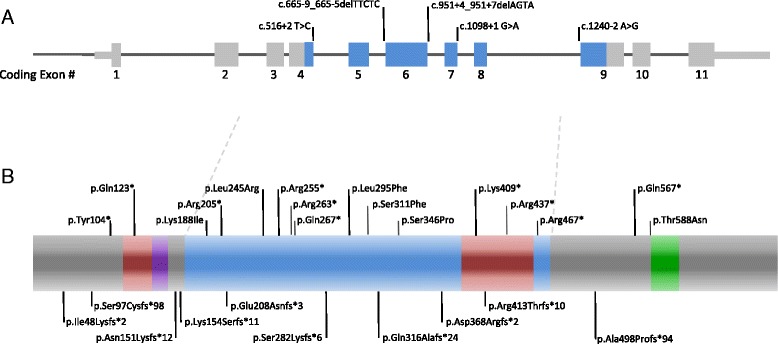
Case presentation: Both individuals presented with the well-known characteristic features, as well as rarer anomalies seen in a minority of patients. Patient 1 presented shortly after birth with an enlarged cisterna magna, distal contractures, and distinctive facies that included bitemporal narrowing and deep set eyes. A de novo splice site mutation in DYRK1A [c.951 + 4_951 + 7delAGTA; p.Val222Aspfs*22] was identified by next generation sequencing. Patient 2 presented at 7 months of age with microcephaly and dysmorphic features. She went several years without a diagnosis until a de novo DYRK1A nonsense mutation [c.787C>T; p.(Arg263*)] was identified at age 12. These individuals, and the 52 cases reviewed from the literature, show the characteristic features of the DYRK1A-related syndrome including global developmental delay, ID, microcephaly, feeding difficulties, and the facial gestalt. Other common findings include seizures, vision defects, brain abnormalities and skeletal abnormalities of the hands and feet. Less common features include optic nerve defects, contractures, ataxia, and cardiac anomalies.
Conclusion: DYRK1A testing should be considered in individuals with the facial features, intellectual disability and post-natal microcephaly. Once diagnosed with DYRK1A-related intellectual disability, a cardiac and ophthalmologic assessment would be recommended as would routine surveillance by a pediatrician for psychomotor development, growth, and feeding.
Figures
Similar articles
-
The DYRK1A gene is a cause of syndromic intellectual disability with severe microcephaly and epilepsy.J Med Genet. 2012 Dec;49(12):731-6. doi: 10.1136/jmedgenet-2012-101251. Epub 2012 Oct 25. J Med Genet. 2012. PMID: 23099646
-
Ten new cases further delineate the syndromic intellectual disability phenotype caused by mutations in DYRK1A.Eur J Hum Genet. 2015 Nov;23(11):1482-7. doi: 10.1038/ejhg.2015.29. Epub 2015 Apr 29. Eur J Hum Genet. 2015. PMID: 25920557 Free PMC article.
-
DYRK1A mutations in two unrelated patients.Eur J Med Genet. 2015 Mar;58(3):168-74. doi: 10.1016/j.ejmg.2014.12.014. Epub 2015 Jan 30. Eur J Med Genet. 2015. PMID: 25641759
-
DYRK1A pathogenic variants in two patients with syndromic intellectual disability and a review of the literature.Mol Genet Genomic Med. 2020 Dec;8(12):e1544. doi: 10.1002/mgg3.1544. Epub 2020 Nov 7. Mol Genet Genomic Med. 2020. PMID: 33159716 Free PMC article. Review.
-
A de novo mutation in RPL10 causes a rare X-linked ribosomopathy characterized by syndromic intellectual disability and epilepsy: A new case and review of the literature.Eur J Med Genet. 2018 Feb;61(2):89-93. doi: 10.1016/j.ejmg.2017.10.011. Epub 2017 Oct 21. Eur J Med Genet. 2018. PMID: 29066376 Review.
Cited by
-
Decoding the Neurodevelopment and Seizure Puzzle: A Pediatric Case of DYRK1A Gene Mutation and Autosomal Dominant Mental Retardation Type 7.Cureus. 2024 Apr 2;16(4):e57460. doi: 10.7759/cureus.57460. eCollection 2024 Apr. Cureus. 2024. PMID: 38566780 Free PMC article.
-
Clinical phenotype of ASD-associated DYRK1A haploinsufficiency.Mol Autism. 2017 Oct 5;8:54. doi: 10.1186/s13229-017-0173-5. eCollection 2017. Mol Autism. 2017. PMID: 29034068 Free PMC article.
-
DCAF7/WDR68 is required for normal levels of DYRK1A and DYRK1B.PLoS One. 2018 Nov 29;13(11):e0207779. doi: 10.1371/journal.pone.0207779. eCollection 2018. PLoS One. 2018. PMID: 30496304 Free PMC article.
-
Structural analysis of pathogenic mutations in the DYRK1A gene in patients with developmental disorders.Hum Mol Genet. 2017 Feb 1;26(3):519-526. doi: 10.1093/hmg/ddw409. Hum Mol Genet. 2017. PMID: 28053047 Free PMC article.
-
Ocular Phenotype Associated with DYRK1A Variants.Genes (Basel). 2021 Feb 5;12(2):234. doi: 10.3390/genes12020234. Genes (Basel). 2021. PMID: 33562844 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
