

Adaptive preconditioning in neurological diseases - therapeutic insights from proteostatic perturbations
- PMID: 26923166
- PMCID: PMC5010532
- DOI: 10.1016/j.brainres.2016.02.033
Adaptive preconditioning in neurological diseases - therapeutic insights from proteostatic perturbations
Abstract
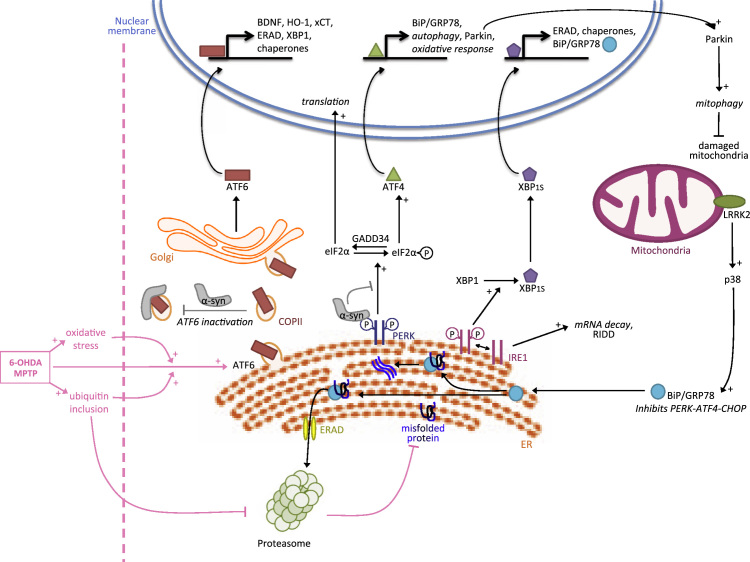
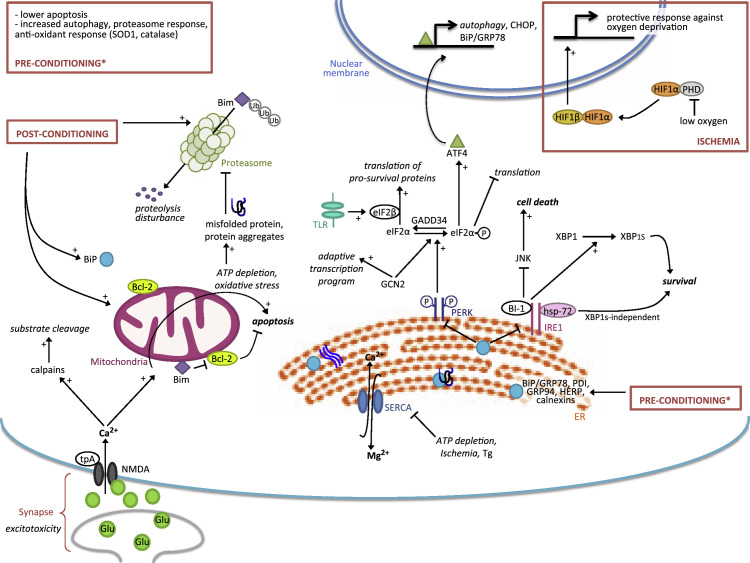
In neurological disorders, both acute and chronic neural stress can disrupt cellular proteostasis, resulting in the generation of pathological protein. However in most cases, neurons adapt to these proteostatic perturbations by activating a range of cellular protective and repair responses, thus maintaining cell function. These interconnected adaptive mechanisms comprise a 'proteostasis network' and include the unfolded protein response, the ubiquitin proteasome system and autophagy. Interestingly, several recent studies have shown that these adaptive responses can be stimulated by preconditioning treatments, which confer resistance to a subsequent toxic challenge - the phenomenon known as hormesis. In this review we discuss the impact of adaptive stress responses stimulated in diverse human neuropathologies including Parkinson׳s disease, Wolfram syndrome, brain ischemia, and brain cancer. Further, we examine how these responses and the molecular pathways they recruit might be exploited for therapeutic gain. This article is part of a Special Issue entitled SI:ER stress.
Keywords: Autophagy; ER stress; Glioblastoma; Hormesis; Ischemia; Parkinson׳s disease; Proteasome; Wolfram syndrome.
Copyright © 2016 The Authors. Published by Elsevier B.V. All rights reserved.
Figures
References
-
- Abisambra J.F., Jinwal U.K., Blair L.J., O׳Leary J.C., 3rd, Li Q., Brady S., Wang L., Guidi C.E., Zhang B., Nordhues B.A., Cockman M., Suntharalingham A., Li P., Jin Y., Atkins C.A., Dickey C.A. Tau accumulation activates the unfolded protein response by impairing endoplasmic reticulum-associated degradation. J. Neurosci. 2013;33:9498–9507. - PMC - PubMed
-
- Arrasate M., Mitra S., Schweitzer E.S., Segal M.R., Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
