

A Stringent Systems Approach Uncovers Gene-Specific Mechanisms Regulating Inflammation
- PMID: 26924576
- PMCID: PMC4808443
- DOI: 10.1016/j.cell.2016.01.020
A Stringent Systems Approach Uncovers Gene-Specific Mechanisms Regulating Inflammation
Abstract
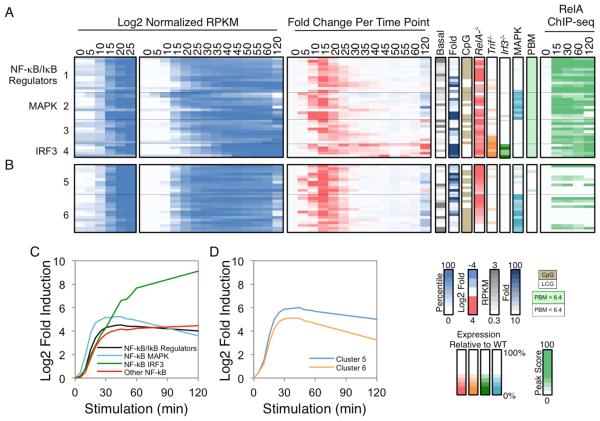
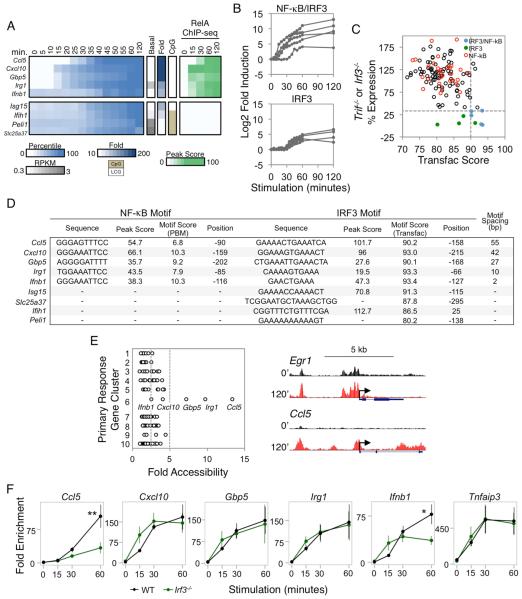
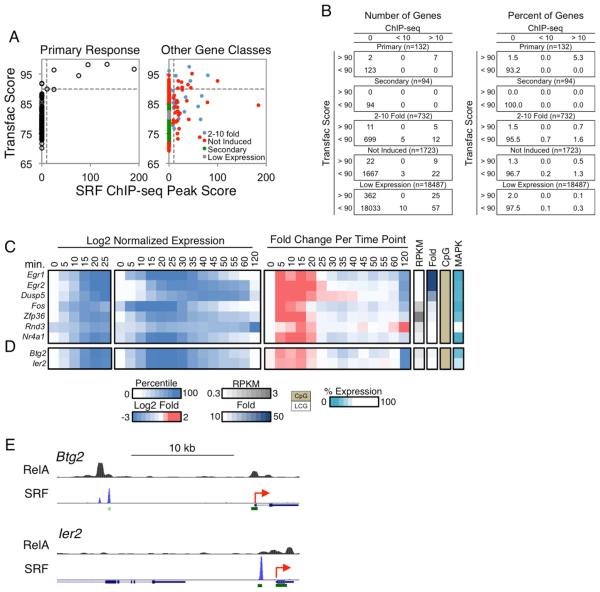
Much has been learned about transcriptional cascades and networks from large-scale systems analyses of high-throughput datasets. However, analysis methods that optimize statistical power through simultaneous evaluation of thousands of ChIP-seq peaks or differentially expressed genes possess substantial limitations in their ability to uncover mechanistic principles of transcriptional control. By examining nascent transcript RNA-seq, ChIP-seq, and binding motif datasets from lipid A-stimulated macrophages with increased attention to the quantitative distribution of signals, we identified unexpected relationships between the in vivo binding properties of inducible transcription factors, motif strength, and transcription. Furthermore, rather than emphasizing common features of large clusters of co-regulated genes, our results highlight the extent to which unique mechanisms regulate individual genes with key biological functions. Our findings demonstrate the mechanistic value of stringent interrogation of well-defined sets of genes as a complement to broader systems analyses of transcriptional cascades and networks.
Copyright © 2016 Elsevier Inc. All rights reserved.
Figures
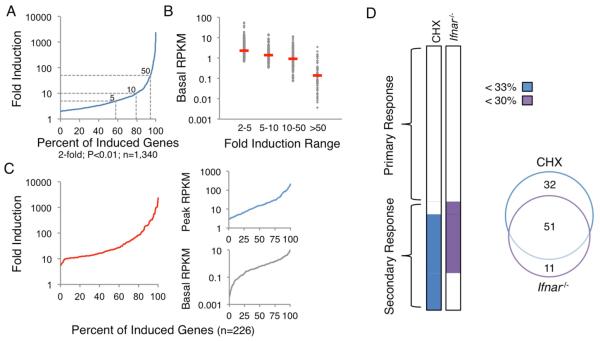
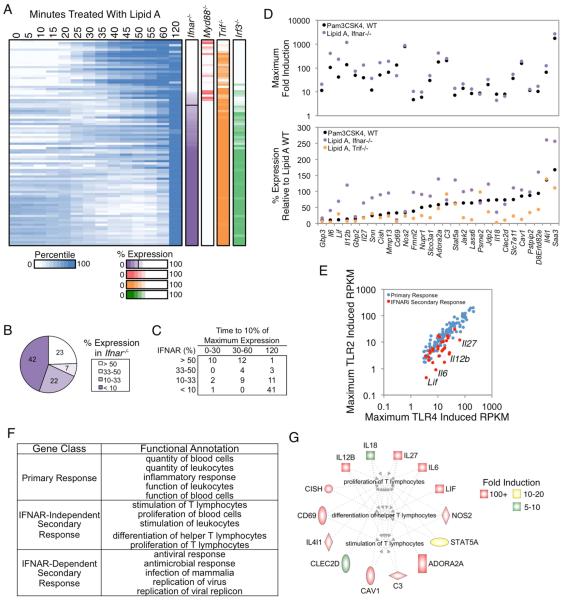
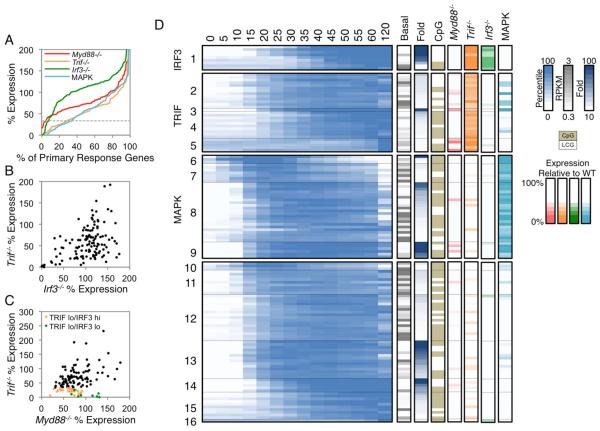
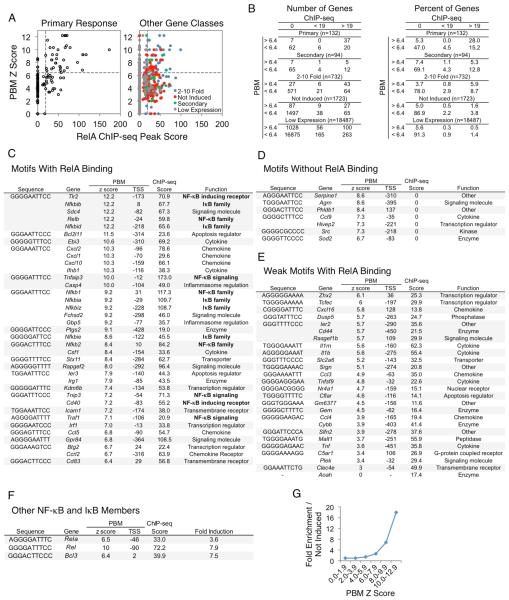
Comment in
-
Gene regulation: A gene-centric analysis of transcriptional cascades.Nat Rev Genet. 2016 Apr;17(4):193. doi: 10.1038/nrg.2016.30. Epub 2016 Mar 7. Nat Rev Genet. 2016. PMID: 26948817 No abstract available.
-
From the Beauty of Genomic Landscapes to the Strength of Transcriptional Mechanisms.Cell. 2016 Mar 24;165(1):18-19. doi: 10.1016/j.cell.2016.03.011. Cell. 2016. PMID: 27015303
References
-
- Agalioti T, Lomvardas S, Parekh B, Yie J, Maniatis T, Thanos D. Ordered Recruitment of Chromatin Modifying and General Transcription Factors to the IFN-β Promoter. Cell. 2000;103:667–678. - PubMed
-
- Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. 1995;57:289–300.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
