

Bacteria within the Gastrointestinal Tract Microbiota Correlated with Improved Growth and Feed Conversion: Challenges Presented for the Identification of Performance Enhancing Probiotic Bacteria
- PMID: 26925052
- PMCID: PMC4760072
- DOI: 10.3389/fmicb.2016.00187
Bacteria within the Gastrointestinal Tract Microbiota Correlated with Improved Growth and Feed Conversion: Challenges Presented for the Identification of Performance Enhancing Probiotic Bacteria
Abstract
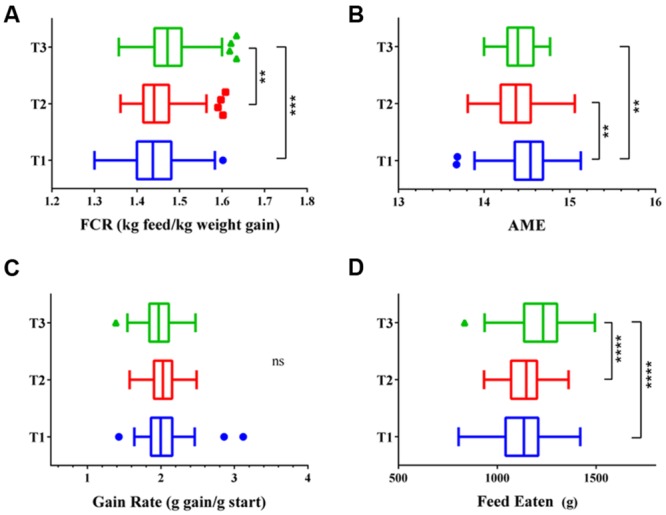
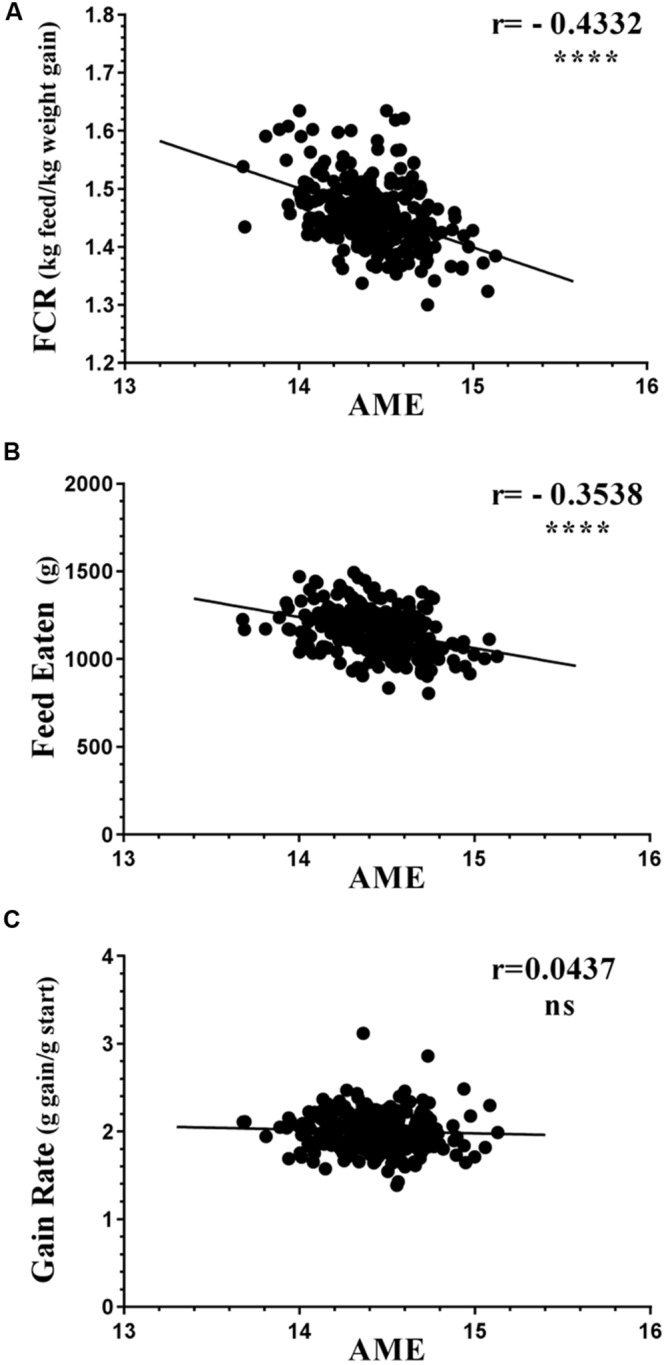
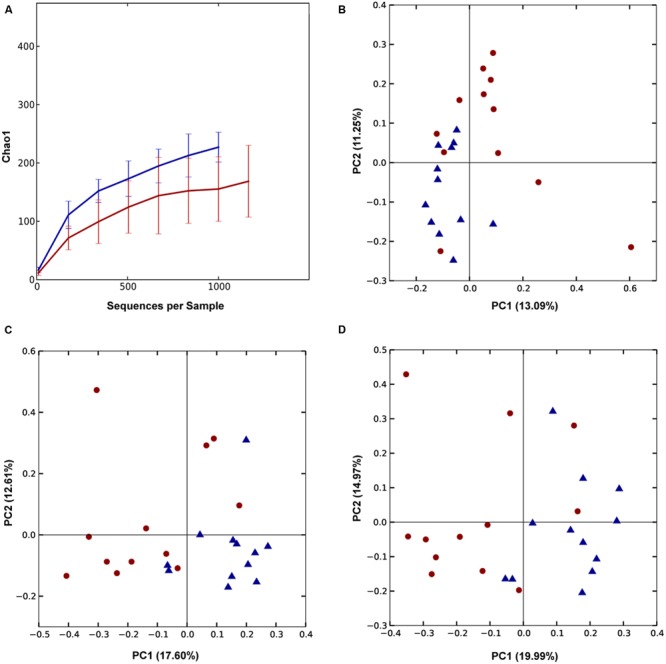
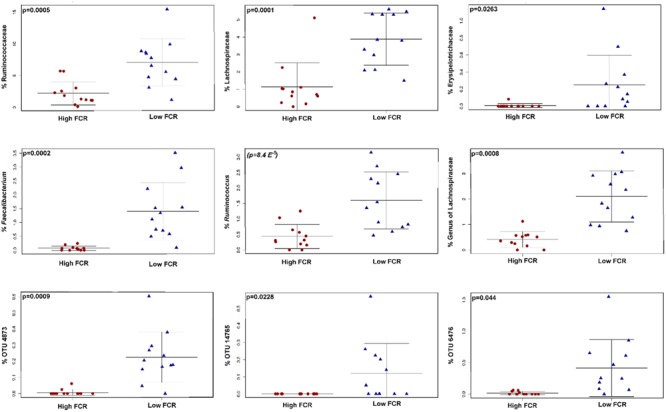
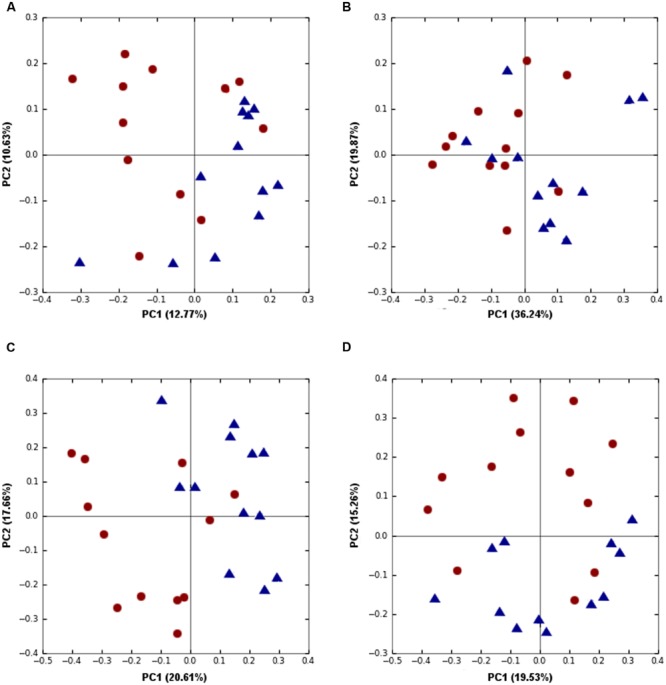
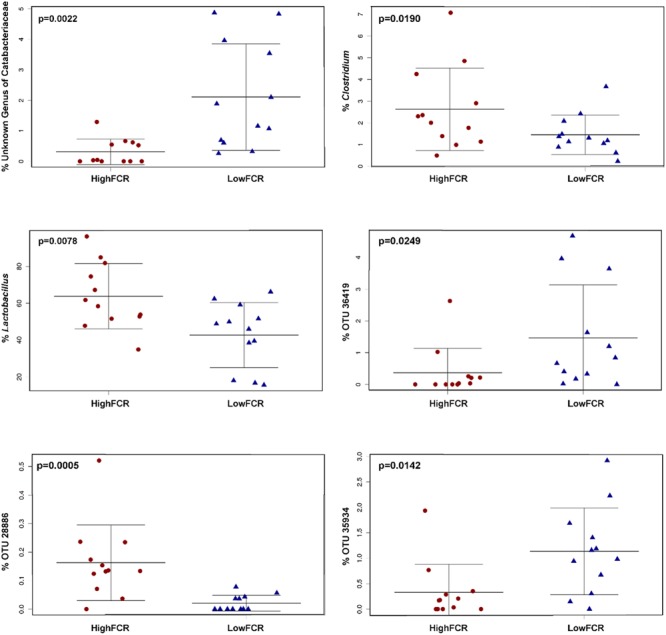
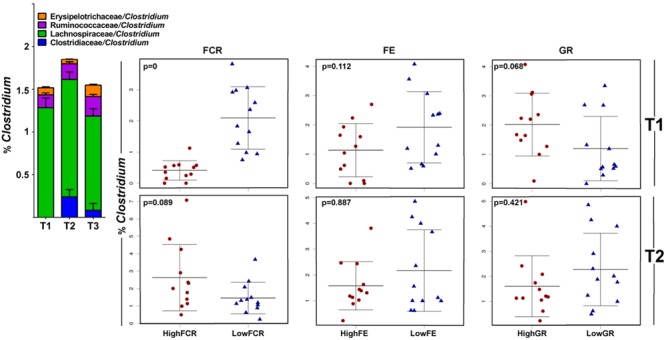
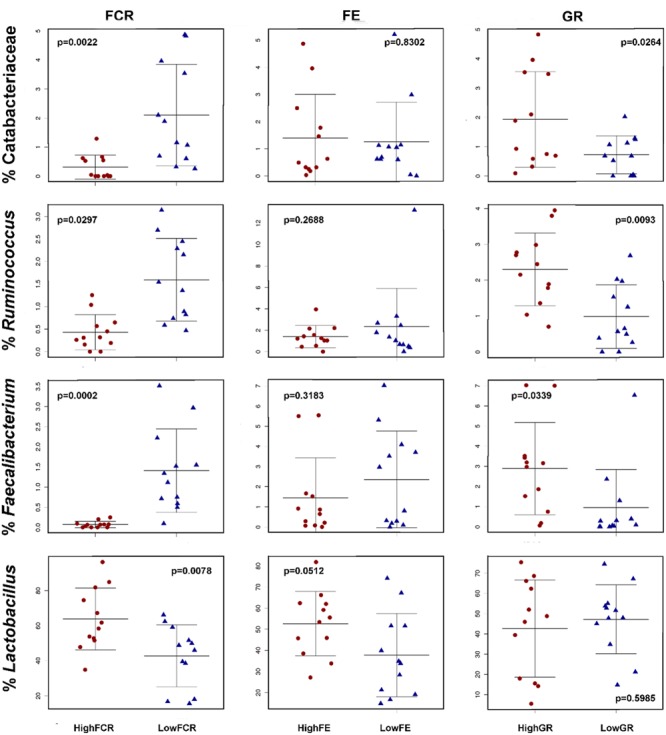
Identification of bacteria associated with desirable productivity outcomes in animals may offer a direct approach to the identification of probiotic bacteria for use in animal production. We performed three controlled chicken trials (n = 96) to investigate caecal microbiota differences between the best and poorest performing birds using four performance measures; feed conversion ratio (FCR), utilization of energy from the feed measured as apparent metabolisable energy, gain rate (GR), and amount of feed eaten (FE). The shifts in microbiota composition associated with the performance measures were very different between the three trials. Analysis of the caecal microbiota revealed that the high and low FCR birds had significant differences in the abundance of some bacteria as demonstrated by shifts in microbiota alpha and beta diversity. Trials 1 and 2 showed significant overall community shifts, however, the microbial changes driving the difference between good and poor performers were very different. Lachnospiraceae, Ruminococcaceae, and Erysipelotrichaceae families and genera Ruminococcus, Faecalibacterium and multiple lineages of genus Clostridium (from families Lachnospiraceae, Ruminococcaceae, and Erysipelotrichaceae) were highly abundant in good FCR birds in Trial 1. Different microbiota was associated with FCR in Trial 2; Catabacteriaceae and unknown Clostridiales family members were increased in good FCR and genera Clostridium (from family Clostridiaceae) and Lactobacillus were associated with poor FCR. Trial 3 had only mild microbiota differences associated with all four performance measures. Overall, the genus Lactobacillus was correlated with feed intake which resulted in poor FCR performance. The genus Faecalibacterium correlated with improved FCR, increased GR and reduced FE. There was overlap in phylotypes correlated with improved FCR and GR, while different microbial cohorts appeared to be correlated with FE. Even under controlled conditions different cohorts of birds developed distinctly different microbiotas. Within the different trial groups the abundance of certain bacterial groups correlated with productivity outcomes. However, with different underlying microbiotas there were different bacteria correlated with performance. The challenge will be to identify probiotic bacteria that can reliably deliver favorable outcomes from diverse microbiotas.
Keywords: caecum; digestive efficiency; energy assimilation; feed conversion; gastrointestinal tract; microbiota; probiotic; weight gain.
Figures
References
-
- Ajuwon K. M. (2015). Toward a better understanding of mechanisms of probiotics and prebiotics in poultry. J. Appl. Poult. Res. 14:2015 10.3382/japr/pfv074 - DOI
-
- Ashelford K. E., Chuzhanova N. A., Fry J. C., Jones A. J., Weightman A. J. (2005). At least 1 in 20 16S rRNA sequence records currently held in public repositories is estimated to contain substantial anomalies. Appl. Environ. Microbiol. 71 7724–7736. 10.1128/AEM.71.12.7724-7736.2005 - DOI - PMC - PubMed
-
- Biddle A., Stewart L., Blanchard J., Leschine S. (2013). Untangling the genetic basis of fibrolytic specialization by lachnospiraceae and ruminococcaceae in diverse gut communities. Diversity 5 627–640. 10.3390/d5030627 - DOI
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases