

Pathogenesis of Nonalcoholic Steatohepatitis
- PMID: 26928243
- PMCID: PMC4887389
- DOI: 10.1053/j.gastro.2016.02.066
Pathogenesis of Nonalcoholic Steatohepatitis
Abstract
Nonalcoholic steatohepatitis (NASH) is a necro-inflammatory response that ensues when hepatocytes are injured by lipids (lipotoxicity). NASH is a potential outcome of nonalcoholic fatty liver (NAFL), a condition that occurs when lipids accumulate in hepatocytes. NASH may be reversible, but it can also result in cirrhosis and primary liver cancer. We are beginning to learn about the mechanisms of progression of NAFL and NASH. NAFL does not inevitably lead to NASH because NAFL is a heterogeneous condition. This heterogeneity exists because different types of lipids with different cytotoxic potential accumulate in the NAFL, and individuals with NAFL differ in their ability to defend against lipotoxicity. There are no tests that reliably predict which patients with NAFL will develop lipotoxicity. However, NASH encompasses the spectrum of wound-healing responses induced by lipotoxic hepatocytes. Differences in these wound-healing responses among individuals determine whether lipotoxic livers regenerate, leading to stabilization or resolution of NASH, or develop progressive scarring, cirrhosis, and possibly liver cancer. We review concepts that are central to the pathogenesis of NASH.
Keywords: Lipotoxicity; Misrepair; Nonalcoholic Fatty Liver Disease; Wound-Healing Response.
Copyright © 2016 AGA Institute. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Figures
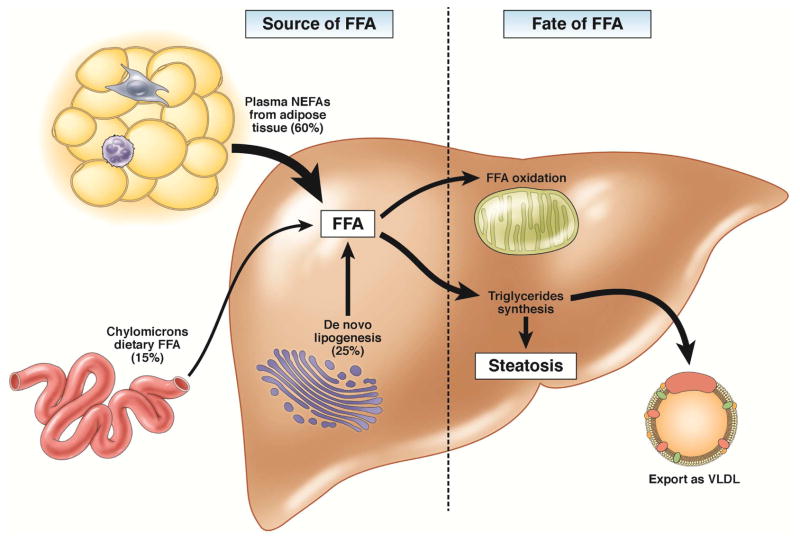
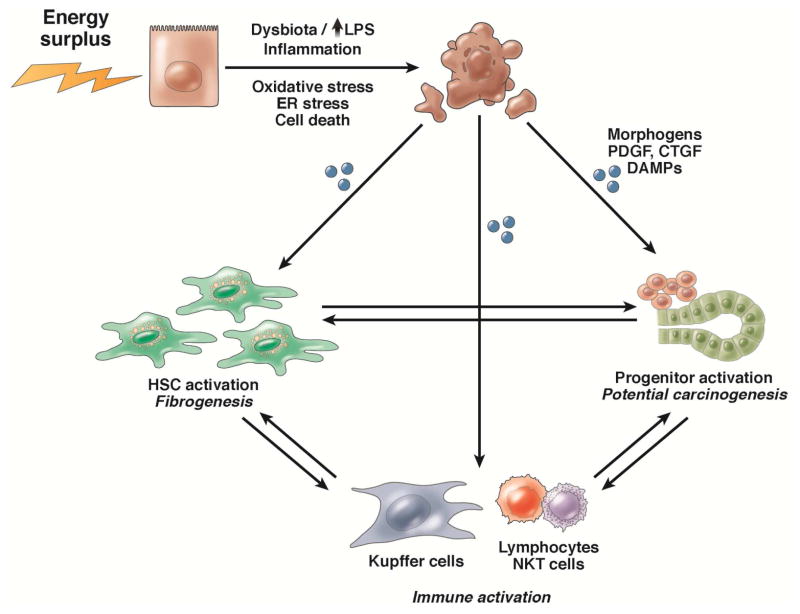
References
-
- Matteoni CA, Younossi ZM, Gramlich T, et al. NAFLD: a spectrum of clinical and pathological severity. Gastroenterology. 1999;116:1413–9. - PubMed
-
- Rafiq N, Bai C, Fang Y, et al. Long-term follow-up of patients with NAFL. Clin Gastroenterol Hepatol. 2009;7:234–8. - PubMed
-
- Musso G, Gambino R, Cassader M, et al. Meta-analysis: natural history of NAFLD and diagnostic accuracy of non-invasive tests for liver disease severity. Ann Med. 2011;43:617–49. - PubMed
-
- Feldstein AE, Canbay A, Angulo P, et al. Hepatocyte apoptosis and fas expression are prominent features of human NASH. Gastroenterology. 2003;125:437–43. - PubMed
-
- Cusi K. Role of obesity and lipotoxicity in the development of NASH: pathophysiology and clinical implications. Gastroenterology. 2012;142:711–725. e6. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
