

ALG1-CDG: Clinical and Molecular Characterization of 39 Unreported Patients
- PMID: 26931382
- PMCID: PMC4907823
- DOI: 10.1002/humu.22983
ALG1-CDG: Clinical and Molecular Characterization of 39 Unreported Patients
Abstract
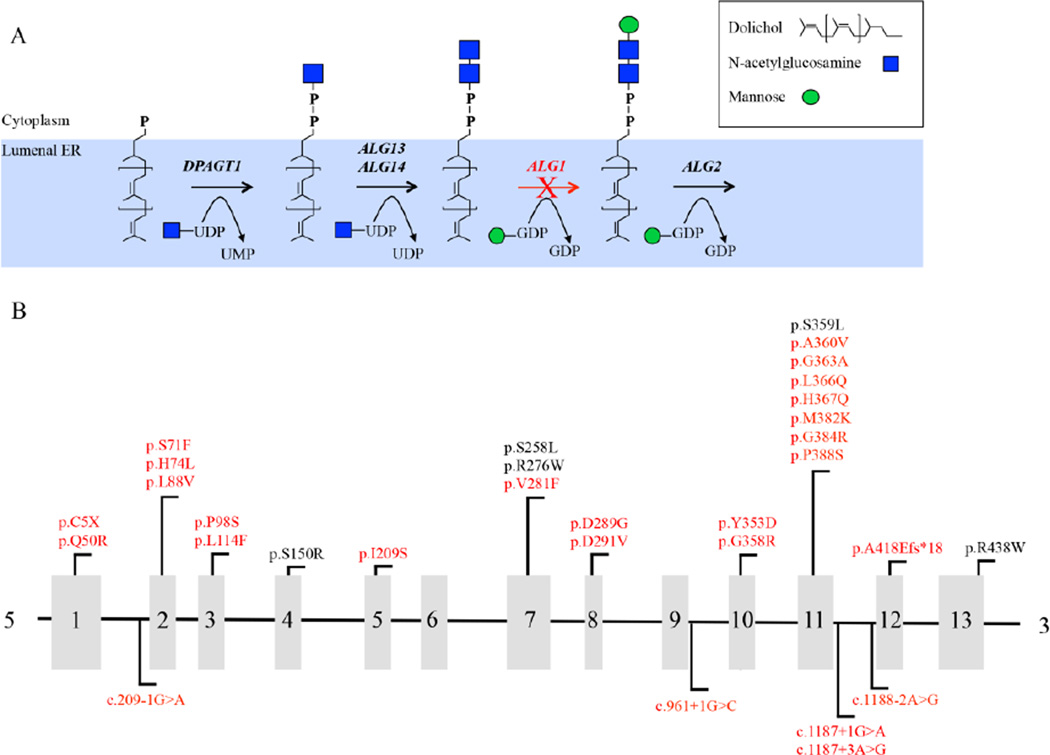
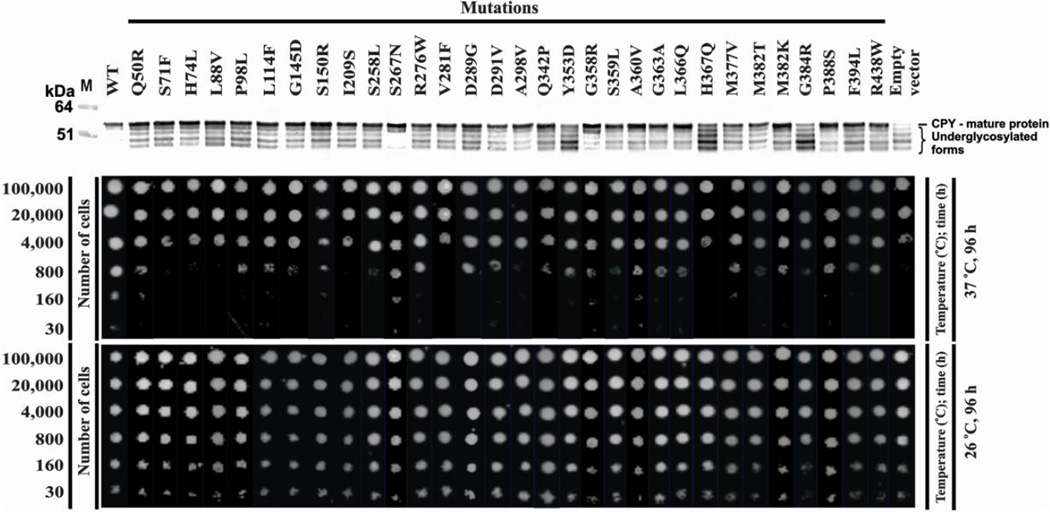
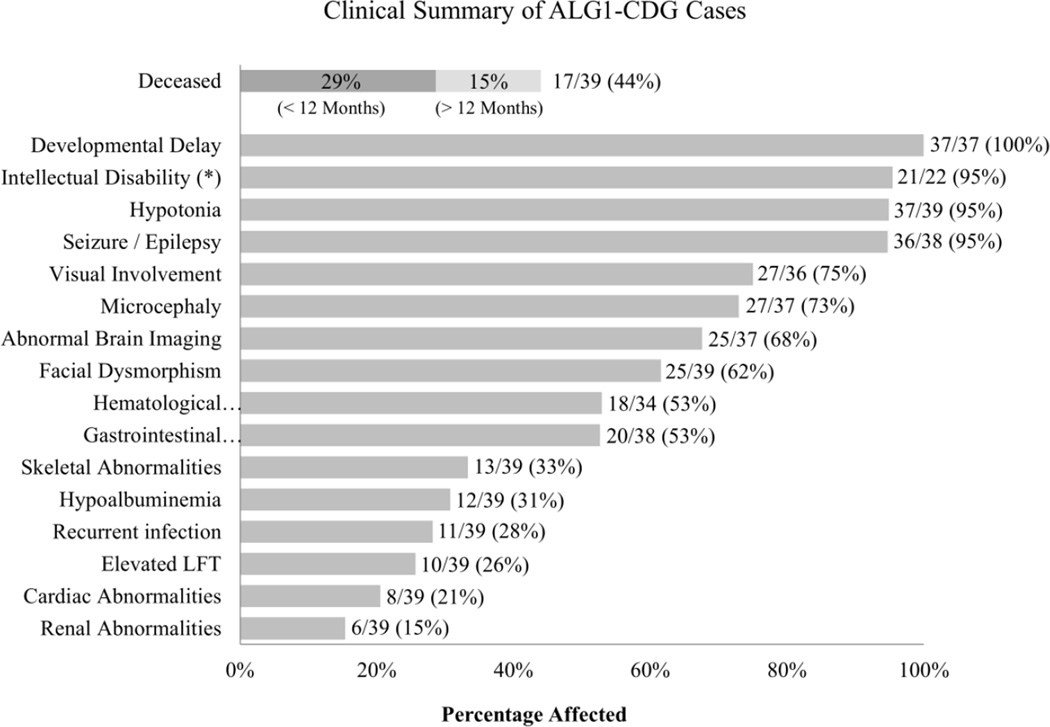
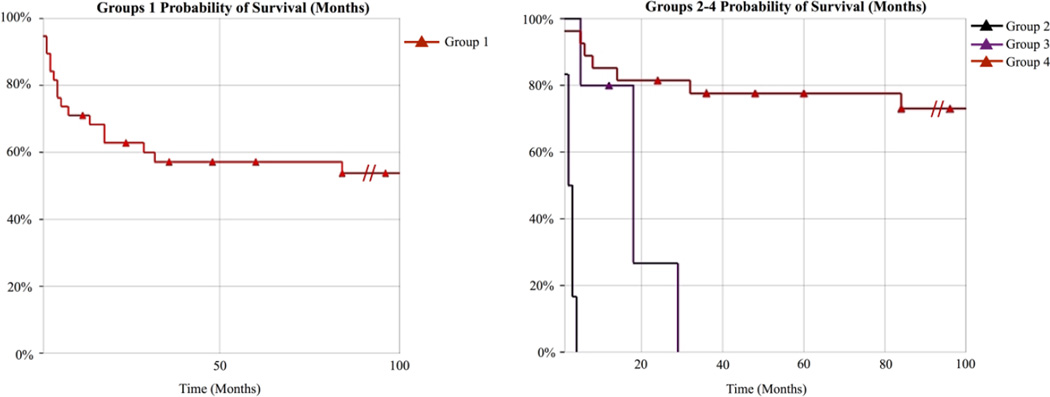
Congenital disorders of glycosylation (CDG) arise from pathogenic mutations in over 100 genes leading to impaired protein or lipid glycosylation. ALG1 encodes a β1,4 mannosyltransferase that catalyzes the addition of the first of nine mannose moieties to form a dolichol-lipid linked oligosaccharide intermediate required for proper N-linked glycosylation. ALG1 mutations cause a rare autosomal recessive disorder termed ALG1-CDG. To date 13 mutations in 18 patients from 14 families have been described with varying degrees of clinical severity. We identified and characterized 39 previously unreported cases of ALG1-CDG from 32 families and add 26 new mutations. Pathogenicity of each mutation was confirmed based on its inability to rescue impaired growth or hypoglycosylation of a standard biomarker in an alg1-deficient yeast strain. Using this approach we could not establish a rank order comparison of biomarker glycosylation and patient phenotype, but we identified mutations with a lethal outcome in the first two years of life. The recently identified protein-linked xeno-tetrasaccharide biomarker, NeuAc-Gal-GlcNAc2 , was seen in all 27 patients tested. Our study triples the number of known patients and expands the molecular and clinical correlates of this disorder.
Keywords: CDG; asparagine-linked glycosylation protein 1; carbohydrate-deficient transferrin; xeno-tetrasaccharide.
© 2016 WILEY PERIODICALS, INC.
Conflict of interest statement
The authors have no conflicts of interest to disclose.
Figures
 ).
).References
-
- Couto JR, Huffaker TC, Robbins PW. Cloning and expression in Escherichia coli of a yeast mannosyltransferase from the asparagine-linked glycosylation pathway. J Biol Chem. 1984;259:378–382. - PubMed
-
- Cueva R, Cotano C, Larriba G. N-glycosylation by transfer of GlcNAc2 from dolichol-PP-GlcNAc2 to the protein moiety of the major yeast exoglucanase. Yeast. 1998;14:773–781. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
