

Drosophila clueless is involved in Parkin-dependent mitophagy by promoting VCP-mediated Marf degradation
- PMID: 26931463
- PMCID: PMC5062585
- DOI: 10.1093/hmg/ddw067
Drosophila clueless is involved in Parkin-dependent mitophagy by promoting VCP-mediated Marf degradation
Erratum in
-
Corrigendum to: Drosophila clueless is involved in Parkin-dependent mitophagy by promoting VCP-mediated Marf degradation.Hum Mol Genet. 2020 Jul 29;29(12):2107. doi: 10.1093/hmg/ddaa092. Hum Mol Genet. 2020. PMID: 32510548 Free PMC article. No abstract available.
Abstract
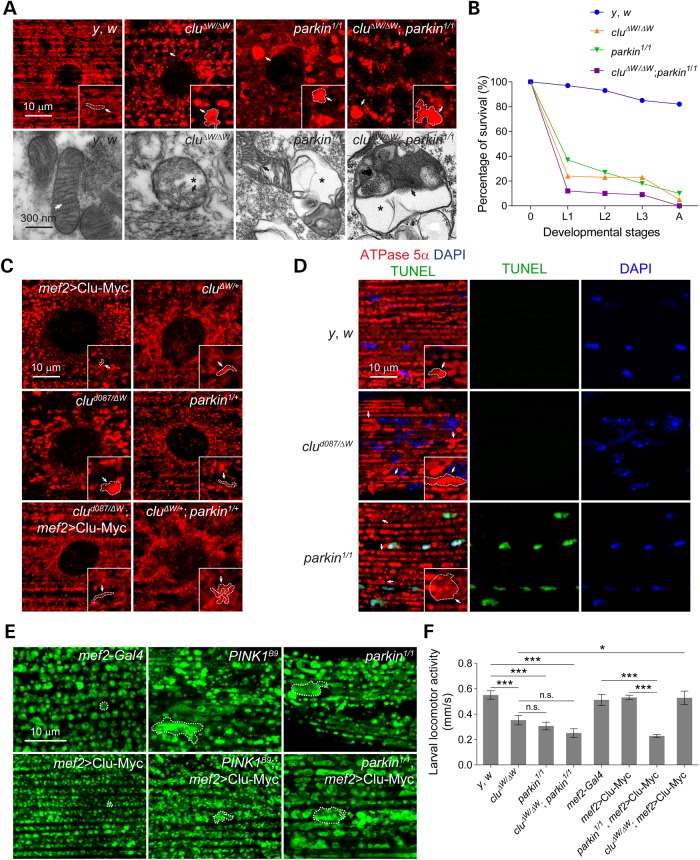
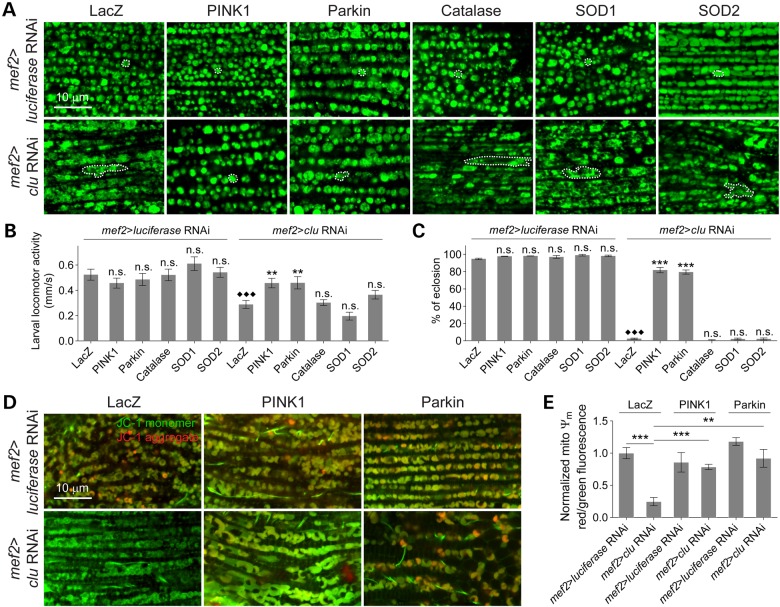
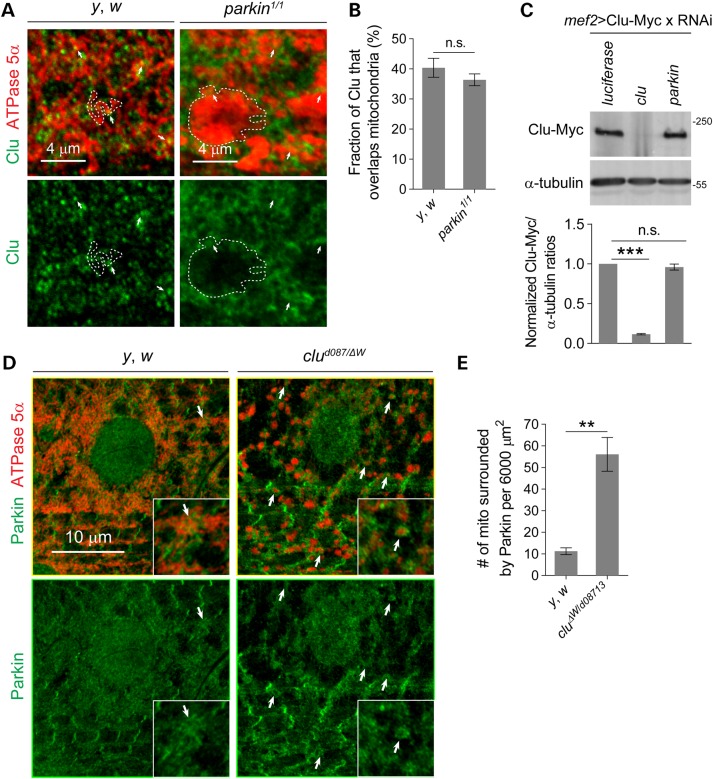
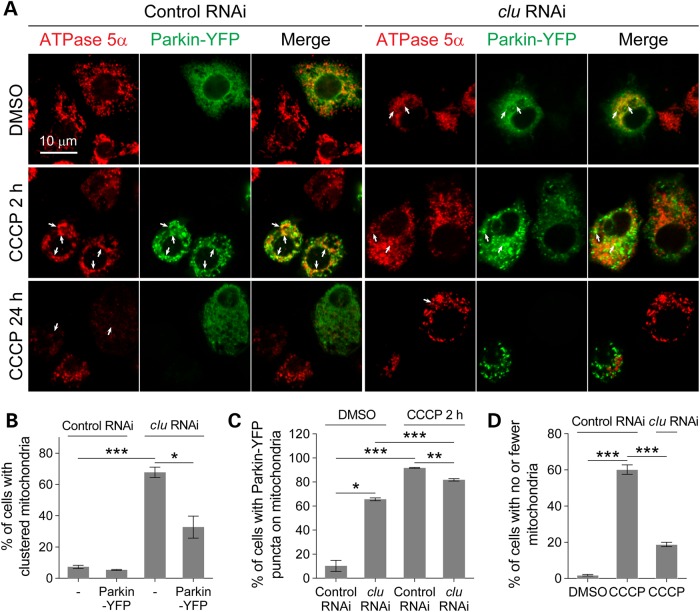
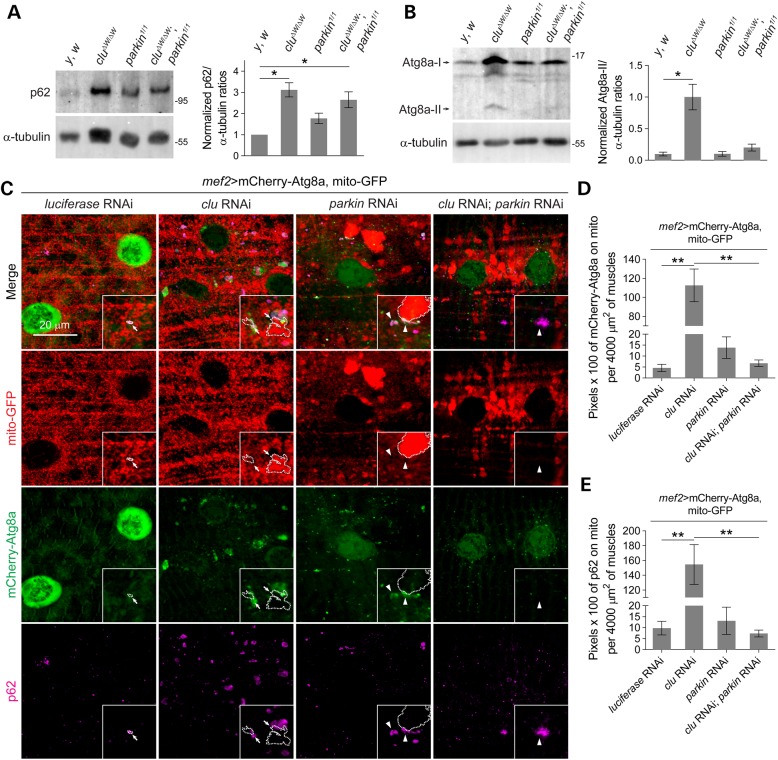
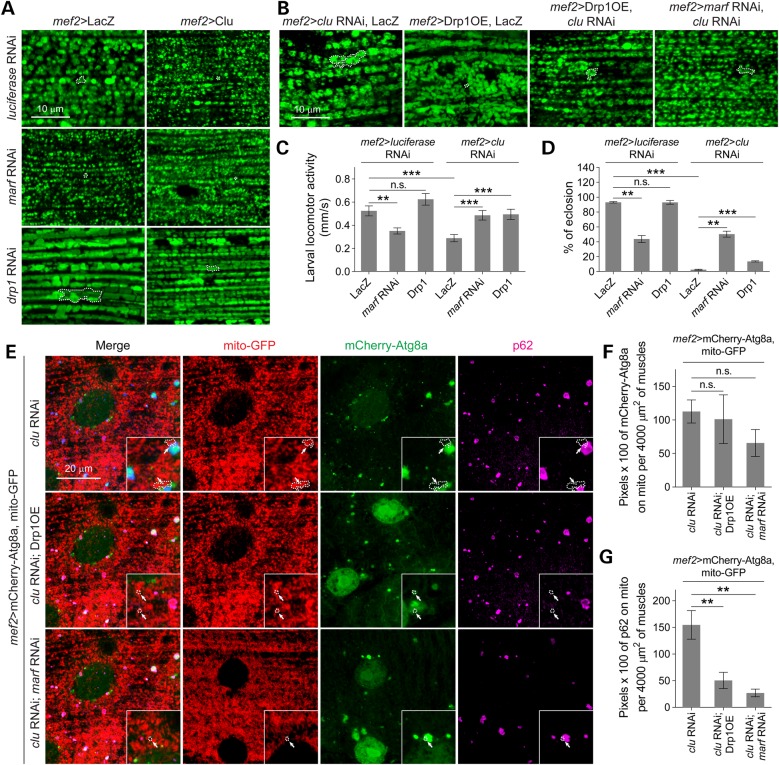
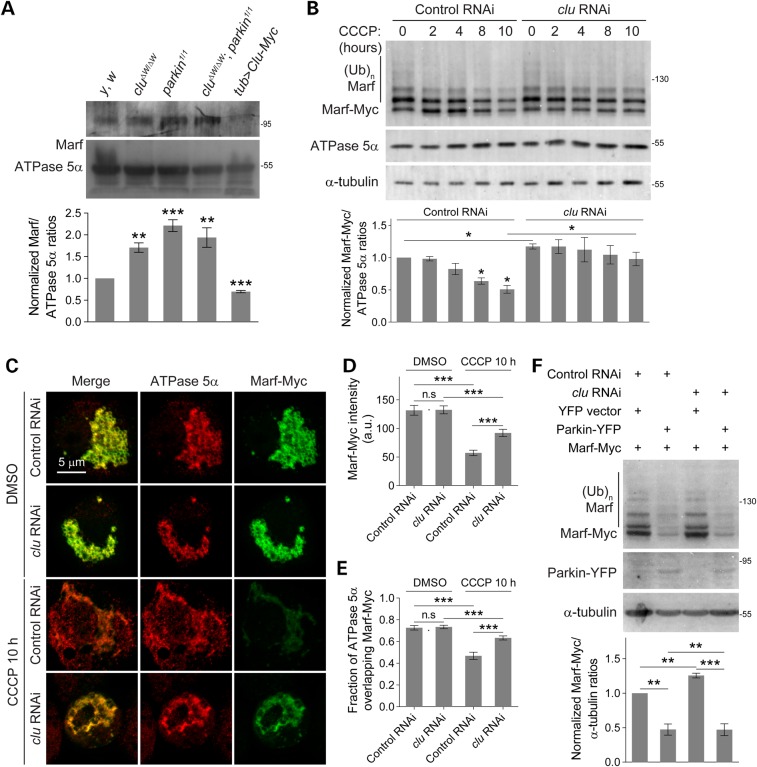
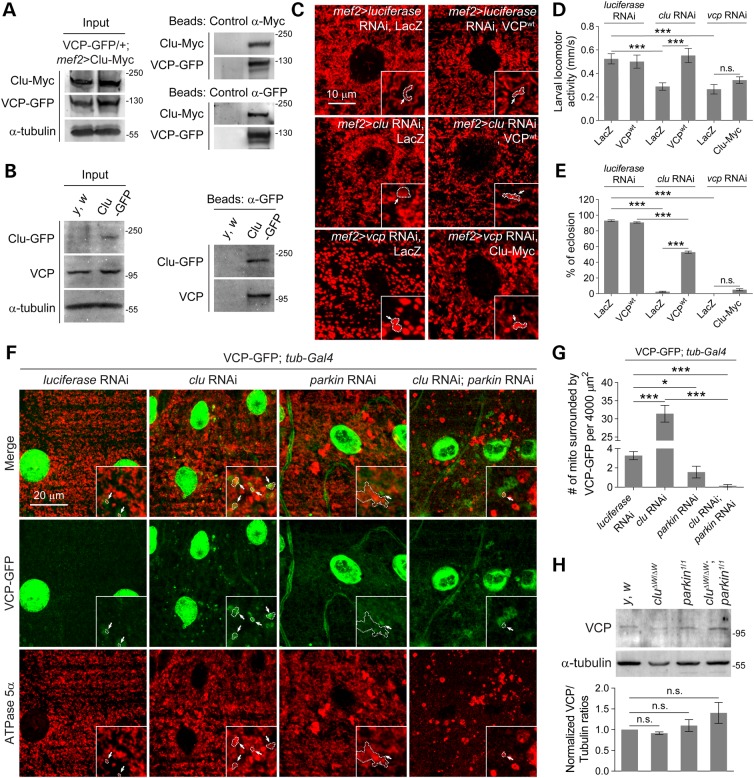
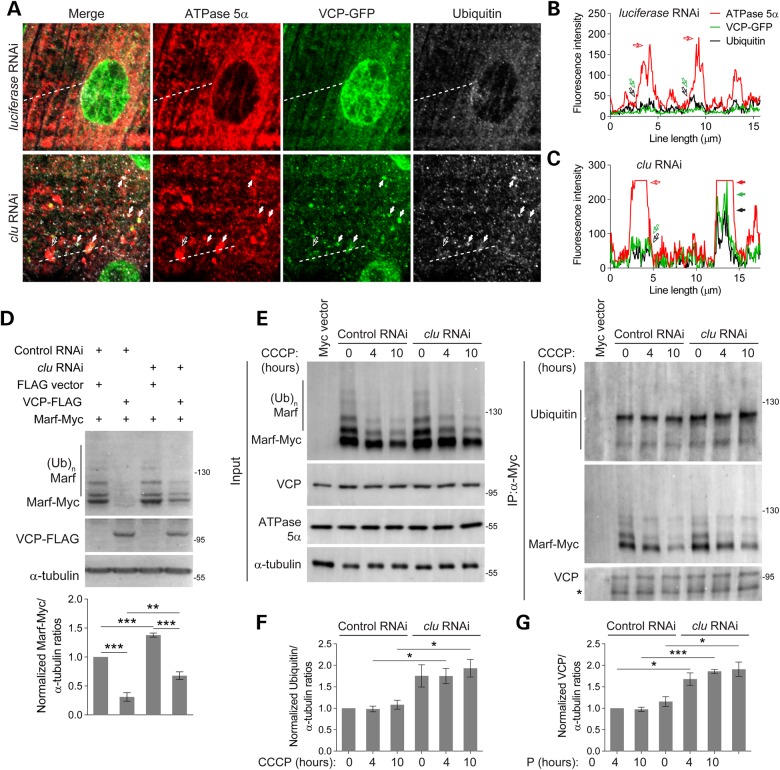
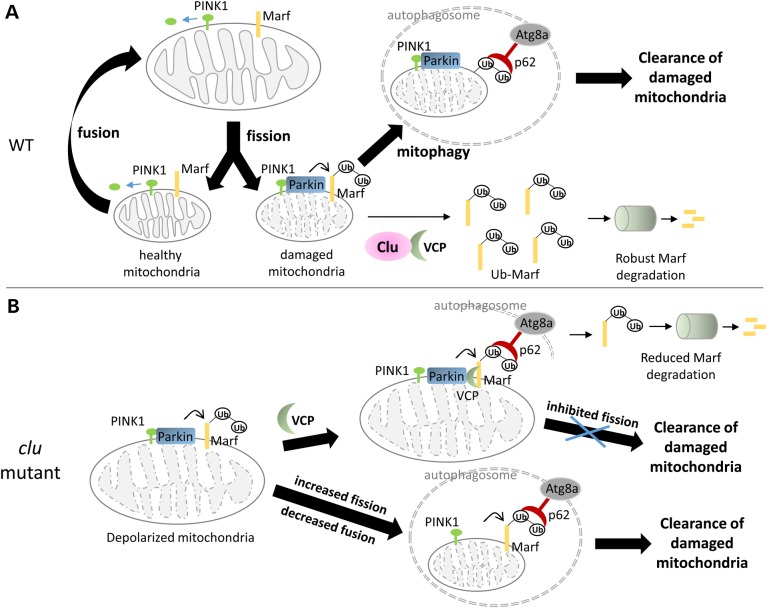
PINK1/Parkin-mediated mitochondrial quality control (MQC) requires valosin-containing protein (VCP)-dependent Mitofusin/Marf degradation to prevent damaged organelles from fusing with the healthy mitochondrial pool, facilitating mitochondrial clearance by autophagy. Drosophila clueless (clu) was found to interact genetically with PINK1 and parkin to regulate mitochondrial clustering in germ cells. However, whether Clu acts in MQC has not been investigated. Here, we show that overexpression of Drosophila Clu complements PINK1, but not parkin, mutant muscles. Loss of clu leads to the recruitment of Parkin, VCP/p97, p62/Ref(2)P and Atg8a to depolarized swollen mitochondria. However, clearance of damaged mitochondria is impeded. This paradox is resolved by the findings that excessive mitochondrial fission or inhibition of fusion alleviates mitochondrial defects and impaired mitophagy caused by clu depletion. Furthermore, Clu is upstream of and binds to VCP in vivo and promotes VCP-dependent Marf degradation in vitro Marf accumulates in whole muscle lysates of clu-deficient flies and is destabilized upon Clu overexpression. Thus, Clu is essential for mitochondrial homeostasis and functions in concert with Parkin and VCP for Marf degradation to promote damaged mitochondrial clearance.
© The Author 2016. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures
Similar articles
-
VCP is essential for mitochondrial quality control by PINK1/Parkin and this function is impaired by VCP mutations.Neuron. 2013 Apr 10;78(1):65-80. doi: 10.1016/j.neuron.2013.02.029. Epub 2013 Mar 14. Neuron. 2013. PMID: 23498974 Free PMC article.
-
Atg1-mediated autophagy suppresses tissue degeneration in pink1/parkin mutants by promoting mitochondrial fission in Drosophila.Mol Biol Cell. 2018 Dec 15;29(26):3082-3092. doi: 10.1091/mbc.E18-04-0243. Epub 2018 Oct 24. Mol Biol Cell. 2018. PMID: 30354903 Free PMC article.
-
Clueless, a protein required for mitochondrial function, interacts with the PINK1-Parkin complex in Drosophila.Dis Model Mech. 2015 Jun;8(6):577-89. doi: 10.1242/dmm.019208. Epub 2015 Apr 20. Dis Model Mech. 2015. PMID: 26035866 Free PMC article.
-
N-degron-mediated degradation and regulation of mitochondrial PINK1 kinase.Curr Genet. 2020 Aug;66(4):693-701. doi: 10.1007/s00294-020-01062-2. Epub 2020 Mar 10. Curr Genet. 2020. PMID: 32157382 Review.
-
Drosophila as a model to study mitochondrial dysfunction in Parkinson's disease.Cold Spring Harb Perspect Med. 2012 Nov 1;2(11):a009944. doi: 10.1101/cshperspect.a009944. Cold Spring Harb Perspect Med. 2012. PMID: 23024178 Free PMC article. Review.
Cited by
-
Mayday sustains trans-synaptic BMP signaling required for synaptic maintenance with age.Elife. 2021 Mar 5;10:e54932. doi: 10.7554/eLife.54932. Elife. 2021. PMID: 33667157 Free PMC article.
-
Clueless forms dynamic, insulin-responsive bliss particles sensitive to stress.Dev Biol. 2020 Mar 15;459(2):149-160. doi: 10.1016/j.ydbio.2019.12.004. Epub 2019 Dec 16. Dev Biol. 2020. PMID: 31837288 Free PMC article.
-
Gp93 safeguards tissue homeostasis by preventing ROS-JNK-mediated apoptosis.Redox Biol. 2025 Apr;81:103537. doi: 10.1016/j.redox.2025.103537. Epub 2025 Feb 8. Redox Biol. 2025. PMID: 39965405 Free PMC article.
-
Loss of nuclear envelope bud formation leads to mitophagy initiation in Drosophila muscles.Autophagy Rep. 2025 Mar 4;4(1):2471121. doi: 10.1080/27694127.2025.2471121. eCollection 2025. Autophagy Rep. 2025. PMID: 40395995 Free PMC article.
-
Disruption of the lipolysis pathway results in stem cell death through a sterile immunity-like pathway in adult Drosophila.Cell Rep. 2022 Jun 21;39(12):110958. doi: 10.1016/j.celrep.2022.110958. Cell Rep. 2022. PMID: 35732115 Free PMC article.
References
-
- Dodson M.W., Guo M. (2007) Pink1, Parkin, DJ-1 and mitochondrial dysfunction in Parkinson's disease. Curr. Opin. Neurobiol., 17, 331–337. - PubMed
-
- Burke W.J. (2002) Recent advances in the genetics and pathogenesis of Parkinson's disease. Neurology, 59, 179–185. - PubMed
-
- Valente E.M., Abou-Sleiman P.M., Caputo V., Muqit M.M., Harvey K., Gispert S., Ali Z., Del Turco D., Bentivoglio A.R., Healy D.G. et al. (2004) Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science, 304, 1158–1160. - PubMed
-
- Kitada T., Asakawa S., Hattori N., Matsumine H., Yamamura Y., Minoshima S., Yokochi M., Mizuno Y., Shimizu N. (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature, 392, 605–608. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
