

Missense variants in the middle domain of DNM1L in cases of infantile encephalopathy alter peroxisomes and mitochondria when assayed in Drosophila
- PMID: 26931468
- PMCID: PMC5007591
- DOI: 10.1093/hmg/ddw059
Missense variants in the middle domain of DNM1L in cases of infantile encephalopathy alter peroxisomes and mitochondria when assayed in Drosophila
Abstract
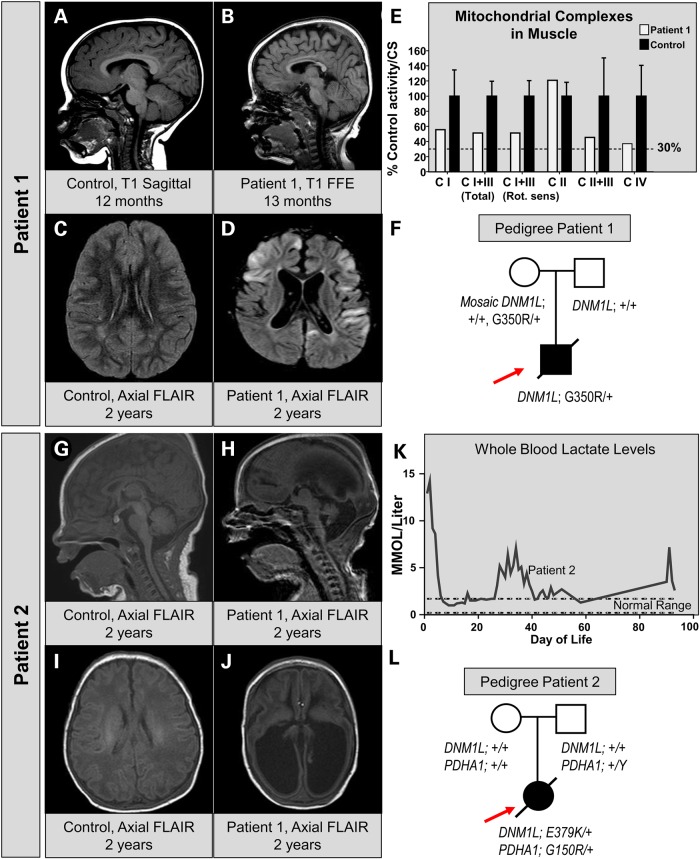
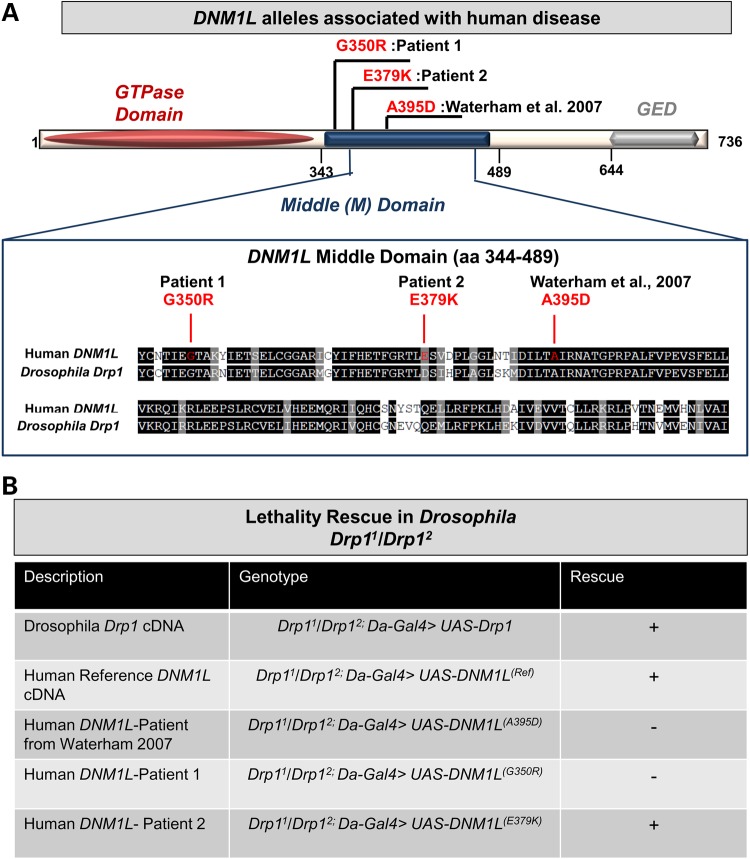
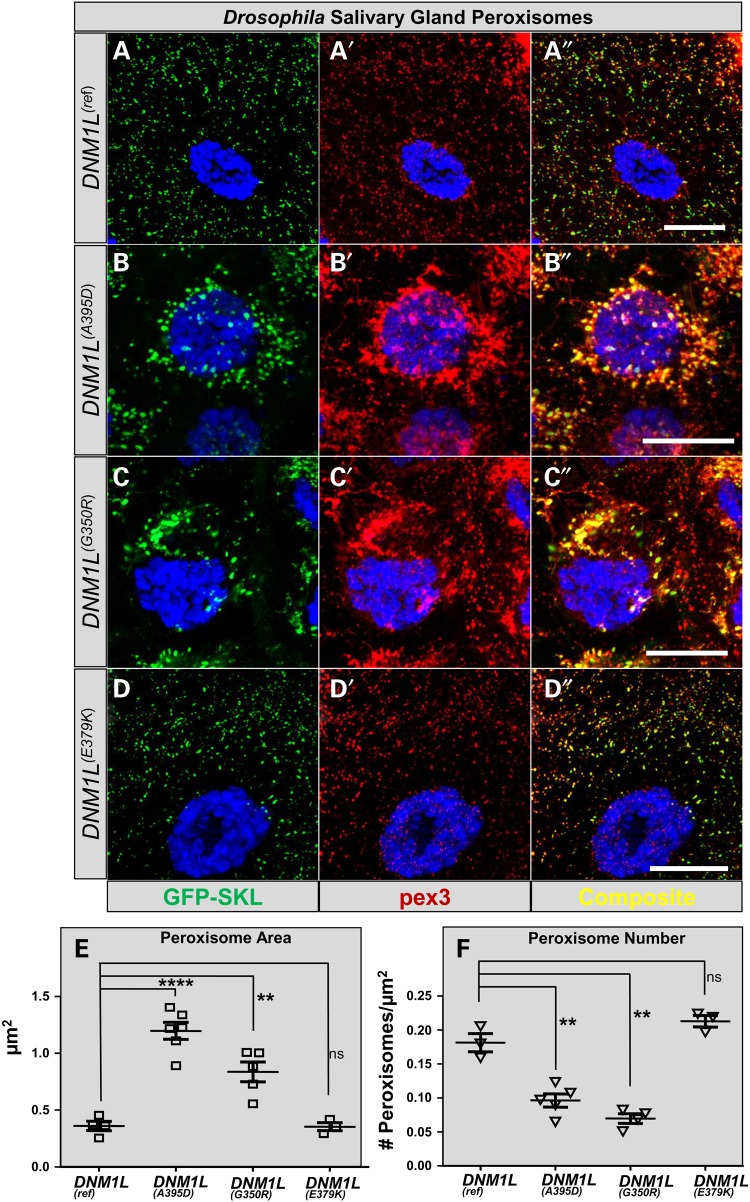
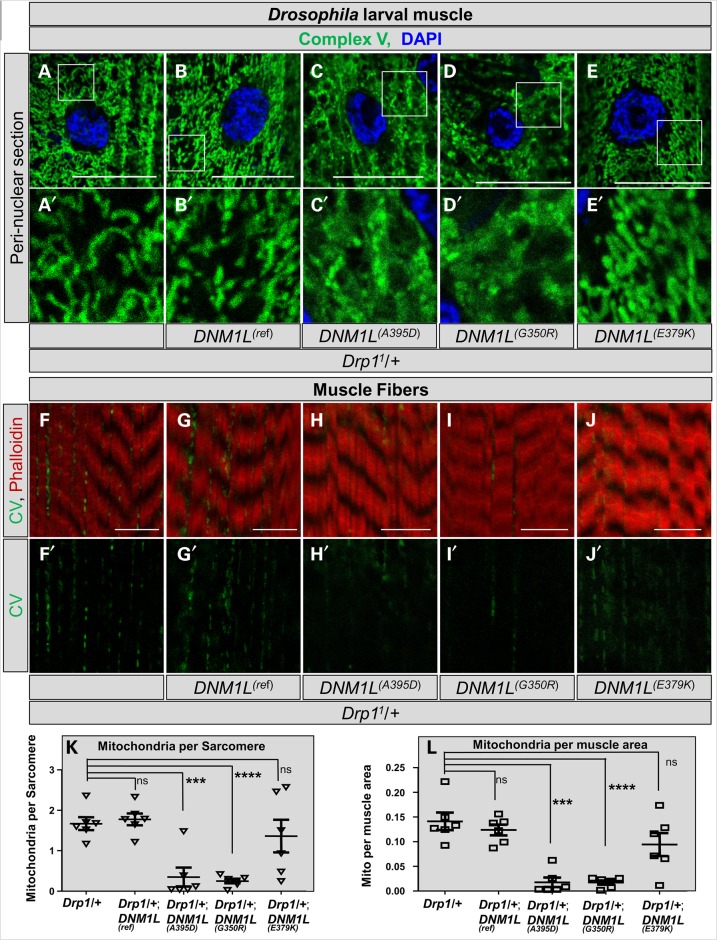
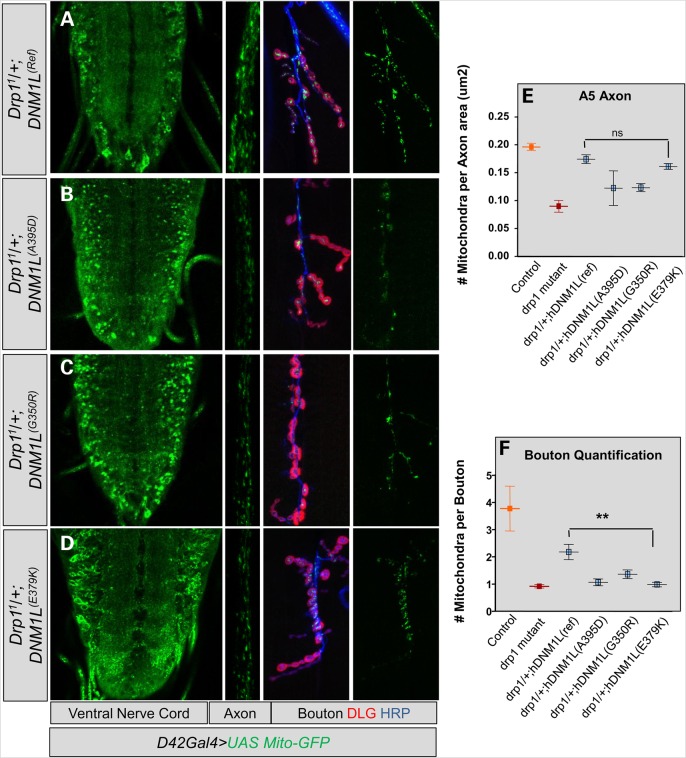
Defects in organelle dynamics underlie a number of human degenerative disorders, and whole exome sequencing (WES) is a powerful tool for studying genetic changes that affect the cellular machinery. WES may uncover variants of unknown significance (VUS) that require functional validation. Previously, a pathogenic de novo variant in the middle domain of DNM1L (p.A395D) was identified in a single patient with a lethal defect of mitochondrial and peroxisomal fission. We identified two additional patients with infantile encephalopathy and partially overlapping clinical features, each with a novel VUS in the middle domain of DNM1L (p.G350R and p.E379K). To evaluate pathogenicity, we generated transgenic Drosophila expressing wild-type or variant DNM1L. We find that human wild-type DNM1L rescues the lethality as well as specific phenotypes associated with the loss of Drp1 in Drosophila. Neither the p.A395D variant nor the novel variant p.G350R rescue lethality or other phenotypes. Moreover, overexpression of p.A395D and p.G350R in Drosophila neurons, salivary gland and muscle strikingly altered peroxisomal and mitochondrial morphology. In contrast, the other novel variant (p.E379K) rescued lethality and did not affect organelle morphology, although it was associated with a subtle mitochondrial trafficking defect in an in vivo assay. Interestingly, the patient with the p.E379K variant also has a de novo VUS in pyruvate dehydrogenase 1 (PDHA1) affecting the same amino acid (G150) as another case of PDHA1 deficiency suggesting the PDHA1 variant may be pathogenic. In summary, detailed clinical evaluation and WES with functional studies in Drosophila can distinguish different functional consequences of newly-described DNM1L alleles.
© The Author 2016. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures
References
-
- Leonard J.V., Schapira A.H. (2000) Mitochondrial respiratory chain disorders I: mitochondrial DNA defects. Lancet, 355, 299–304. - PubMed
-
- Leonard J.V., Schapira A.H. (2000) Mitochondrial respiratory chain disorders II: neurodegenerative disorders and nuclear gene defects. Lancet, 355, 389–394. - PubMed
-
- Lombes A., Aure K., Bellanne-Chantelot C., Gilleron M., Jardel C. (2014) Unsolved issues related to human mitochondrial diseases. Biochimie, 100, 171–176. - PubMed
-
- Tuppen H.A., Blakely E.L., Turnbull D.M., Taylor R.W. (2010) Mitochondrial DNA mutations and human disease. Biochim. Biophys. Acta, 1797, 113–128. - PubMed
MeSH terms
Substances
Supplementary concepts
Grants and funding
- UC2 HL103010/HL/NHLBI NIH HHS/United States
- RC2 HL102926/HL/NHLBI NIH HHS/United States
- K08 NS076547/NS/NINDS NIH HHS/United States
- RC2 HL102924/HL/NHLBI NIH HHS/United States
- RC2 HL103010/HL/NHLBI NIH HHS/United States
- R01 HG011795/HG/NHGRI NIH HHS/United States
- RC2 HL102923/HL/NHLBI NIH HHS/United States
- UC2 HL102926/HL/NHLBI NIH HHS/United States
- UC2 HL102923/HL/NHLBI NIH HHS/United States
- UC2 HL102924/HL/NHLBI NIH HHS/United States
- RC2 HL102925/HL/NHLBI NIH HHS/United States
- UC2 HL102925/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
